Structure, Bonding and Adaptive Aromaticity in Rhenium-oxo Complexes: A DFT Study
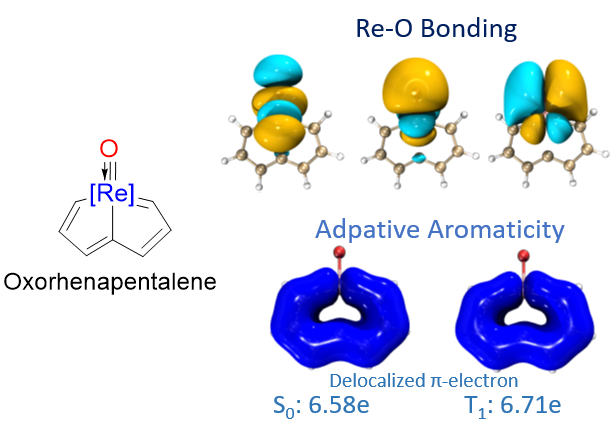
In general, species could be aromatic in the ground or excited state only according to Hückel’s and Baird’s rules. Thus, adaptive aromatics are particularly rare as they can be aromatic in both the lowest singlet and triplet states (S0 and T1). Here, we carry out density functional theory calculations on a series of rhenium oxo complexes to examine their structure, bonding, and aromaticity. It is found that all these complexes are aromatic in the S0 state. In contrast, their T1 states could be antiaromatic, nonaromatic, or aromatic depending on the ligands. Further study suggests that it is the spin electron (de)localization that determines the (anti)aromaticity in their T1 states. Our findings expand the scope of adaptive aromaticity.
https://pubs.rsc.org/en/content/articlelanding/2022/nj/d2nj00911k