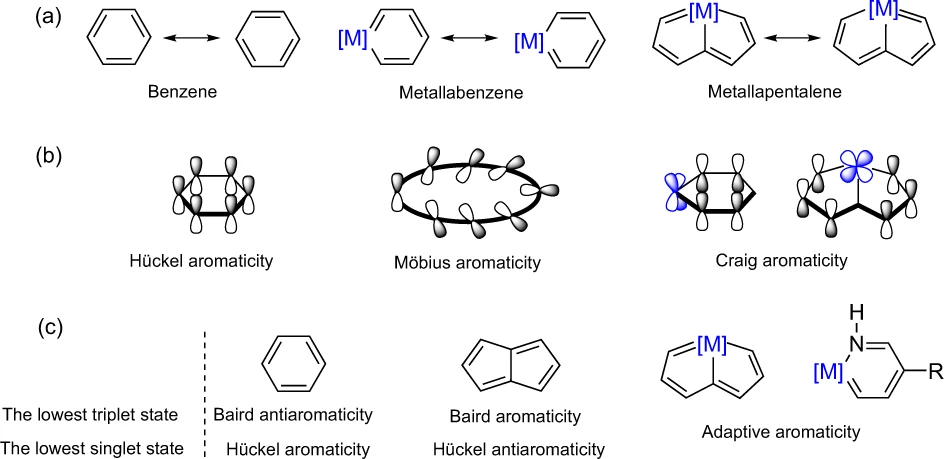
Probing the Tautomerization of Disilenes, Disilabenzenes with Their Isomeric Silylenes: Significant Substituent, Aromaticity and Base Effects
Submitted by Jun Zhu on Thu, 11/12/2020 - 19:24
Disilene has attracted considerable interests due to the trans-bending geometry which is significantly different from the planar alkene. However, the equilibrium between disilene and isomeric silylsilylene has not been fully understood. Here, we report a density functional theory (DFT) study on this equilibrium. Calculations reveal significant effects of substituent, aromaticity and base. Specifically, the parent disilene is thermodynamically more stable than the isomeric silylene.