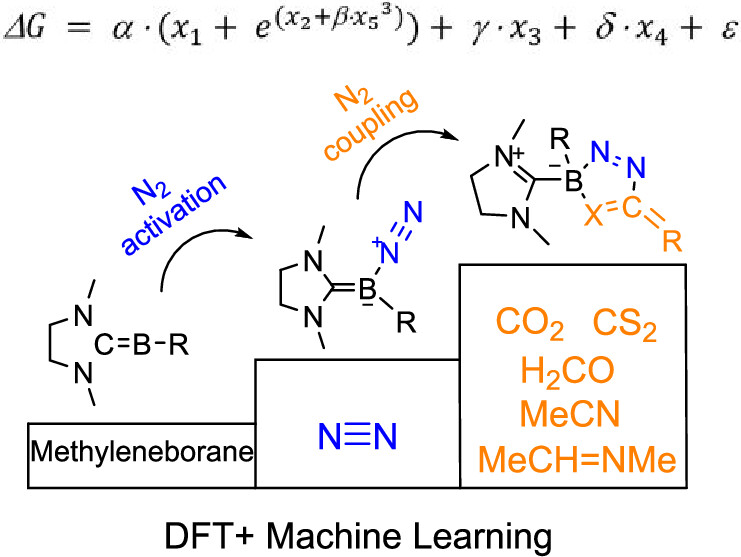
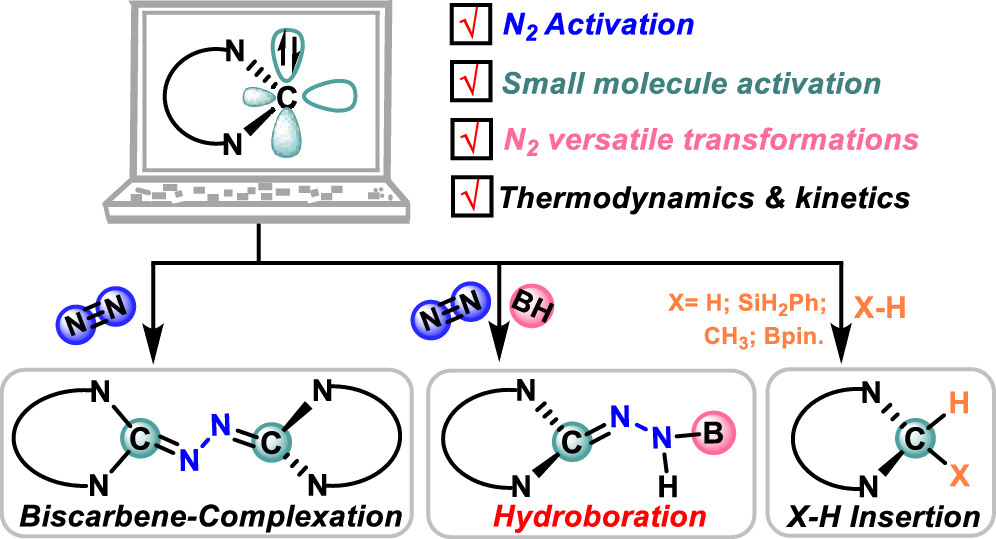
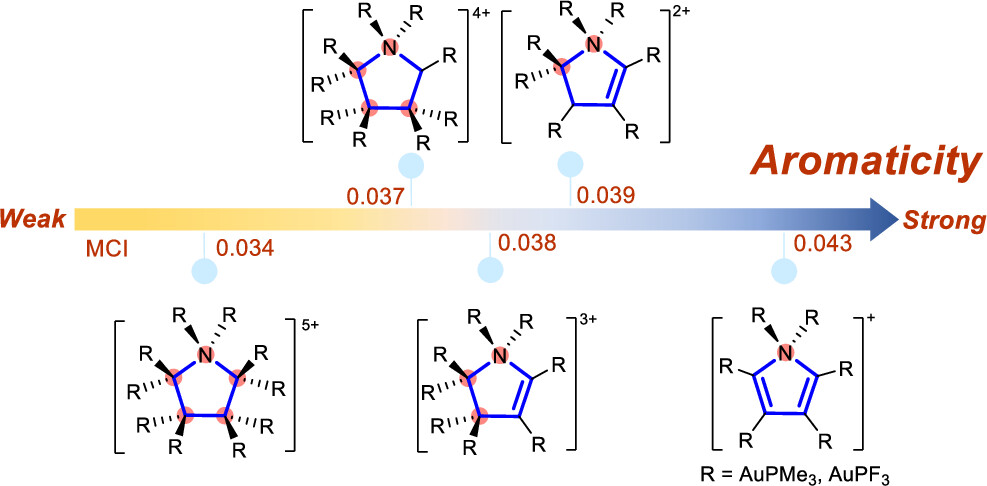
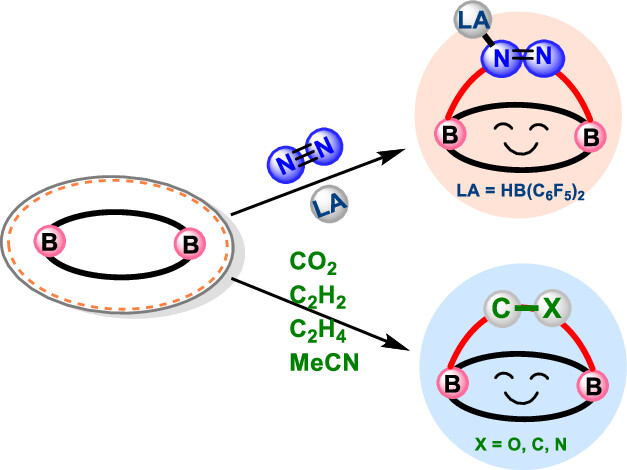
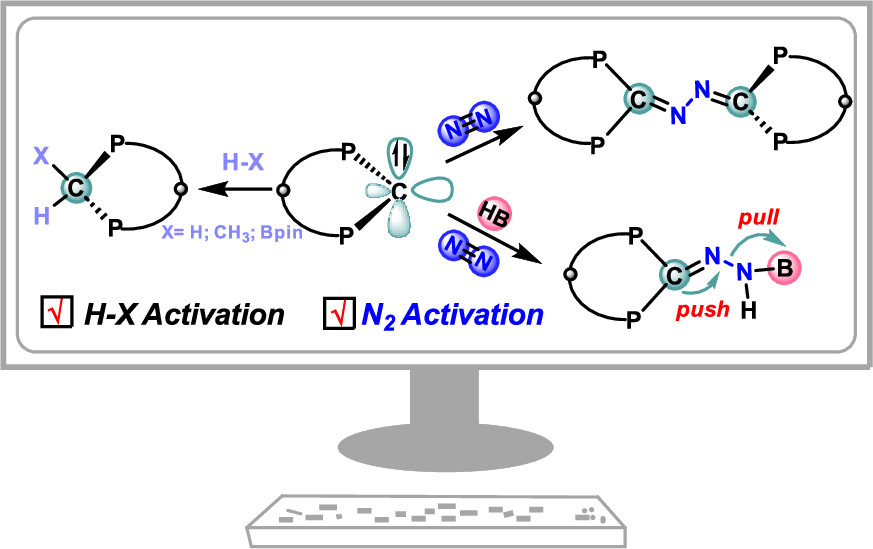
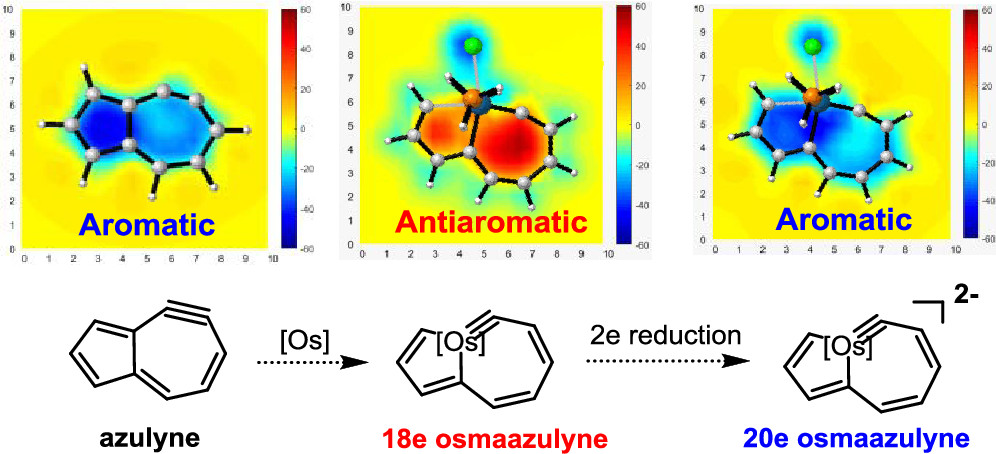
Design of Borole- and Silylene-Based Frustrated Lewis Pairs for Dinitrogen Activation Promoted by Aromaticity: A Combined DFT and Machine Learning Study
Submitted by Jun Zhu on Thu, 09/04/2025 - 09:40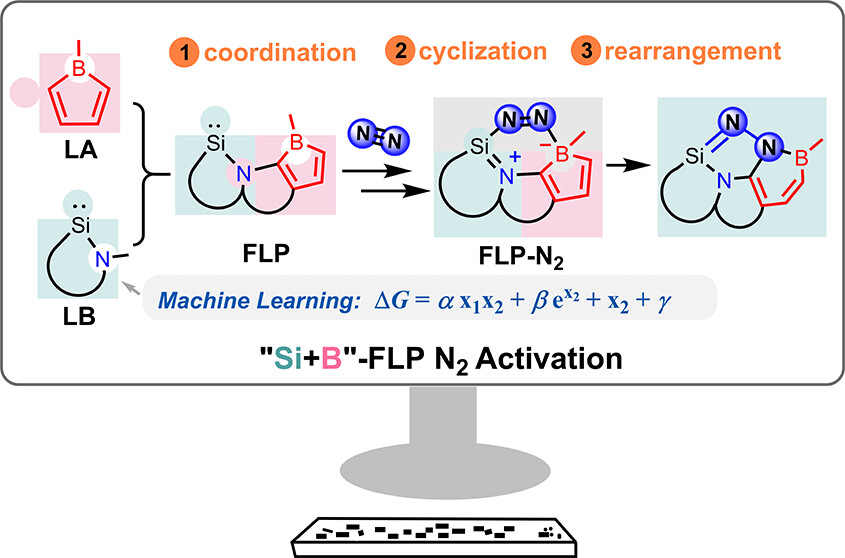
In recent years, while main-group elements from the s- and p-block have emerged in the field of N2 activation, silylenes─despite their remarkable successes in the activation of diverse small molecules─remain unreported for N2 activation. Herein, we design “silylene-borole” frustrated Lewis pairs (FLPs) by combining silylene moieties with boron components and conduct comprehensive density functional theory (DFT) calculations to thoroughly investigate their potential for N2 activation.