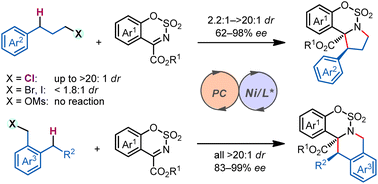
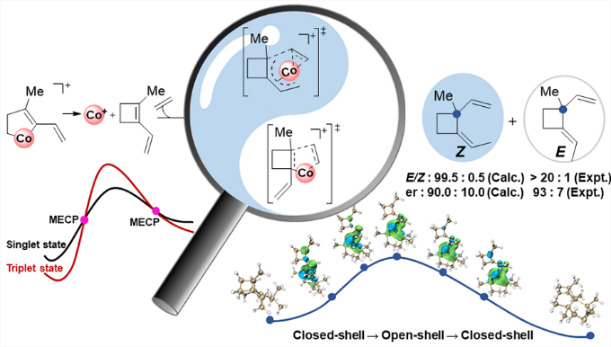
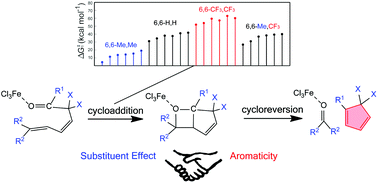
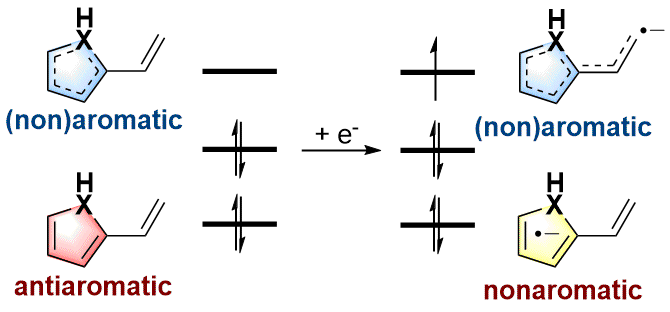"Sacrificial reagent" strategy for tailoring adaptive aromaticity in pyrrole rings with its application to singlet fission: a combined DFT and machine learning study
Submitted by Jun Zhu on Mon, 11/03/2025 - 15:21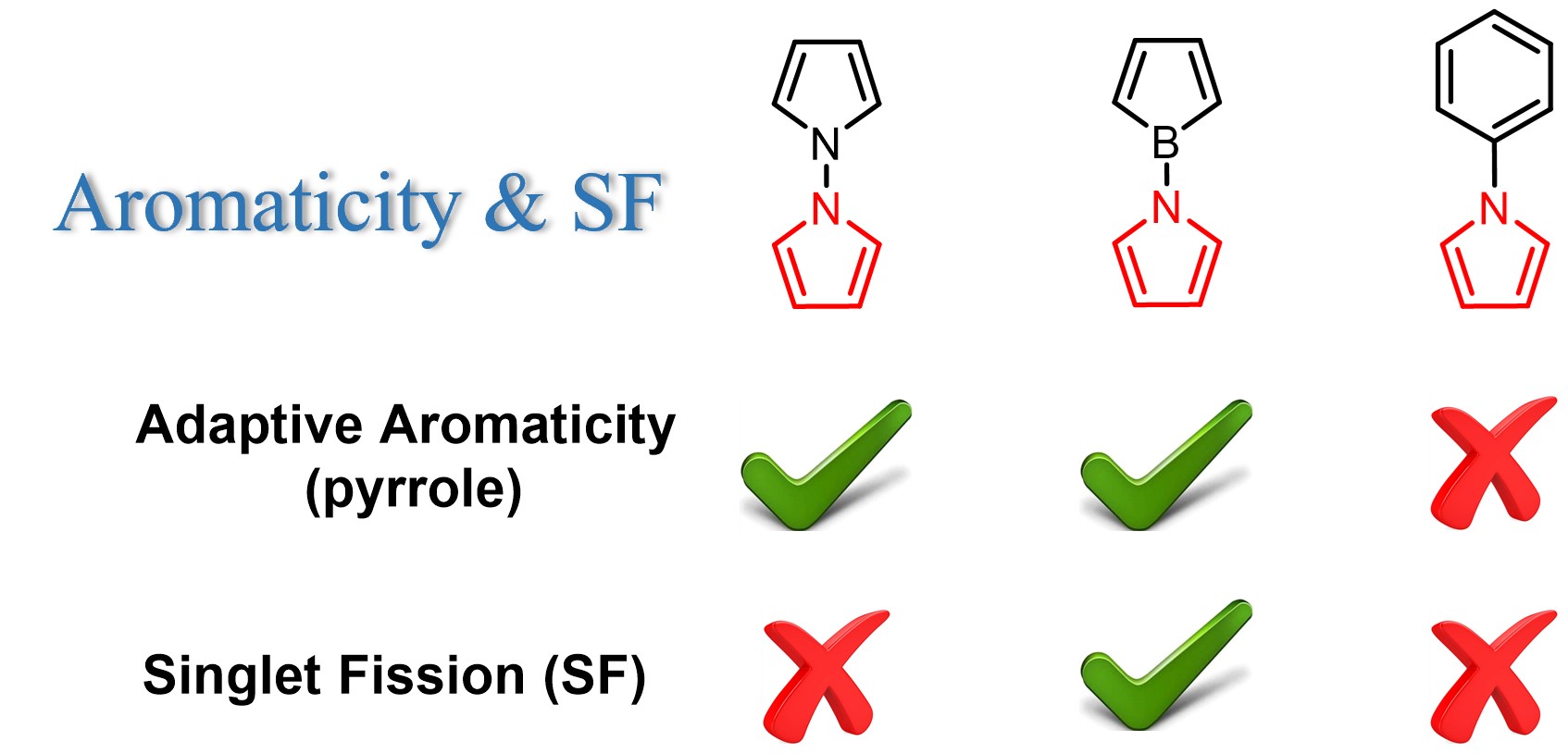
Adaptive aromaticity has attracted substantial attention due to its unconventional two-state aromaticity in both the lowest singlet state (S0) and the lowest triplet state (T1) and its application to the singlet fission. Here we carry out a comprehensive investigation on a series of pyrrole derivatives by density functional theory (DFT) calculations and machine learning.