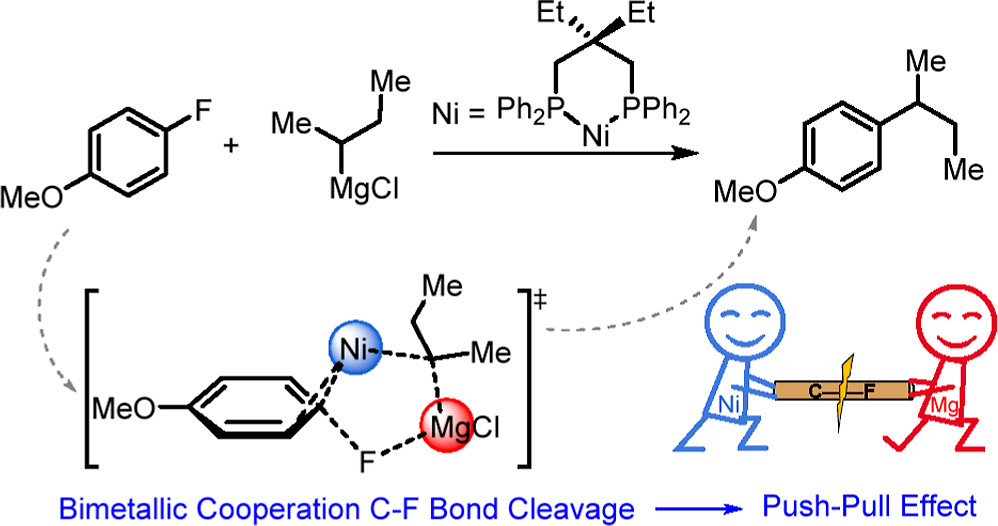
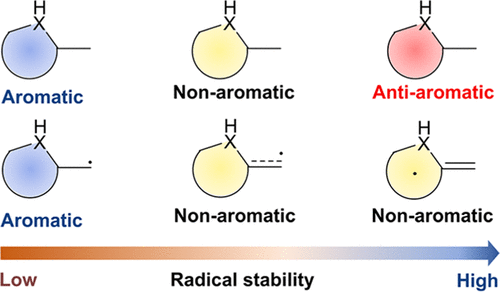
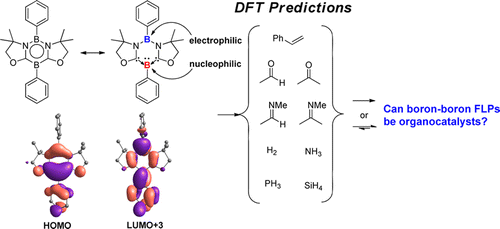
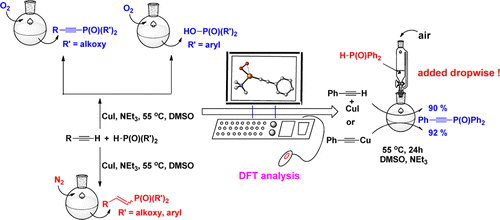
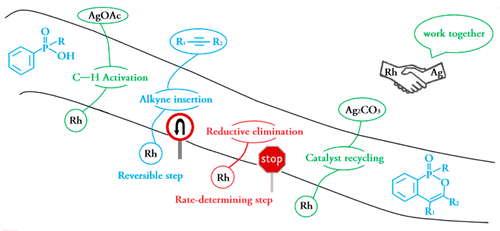
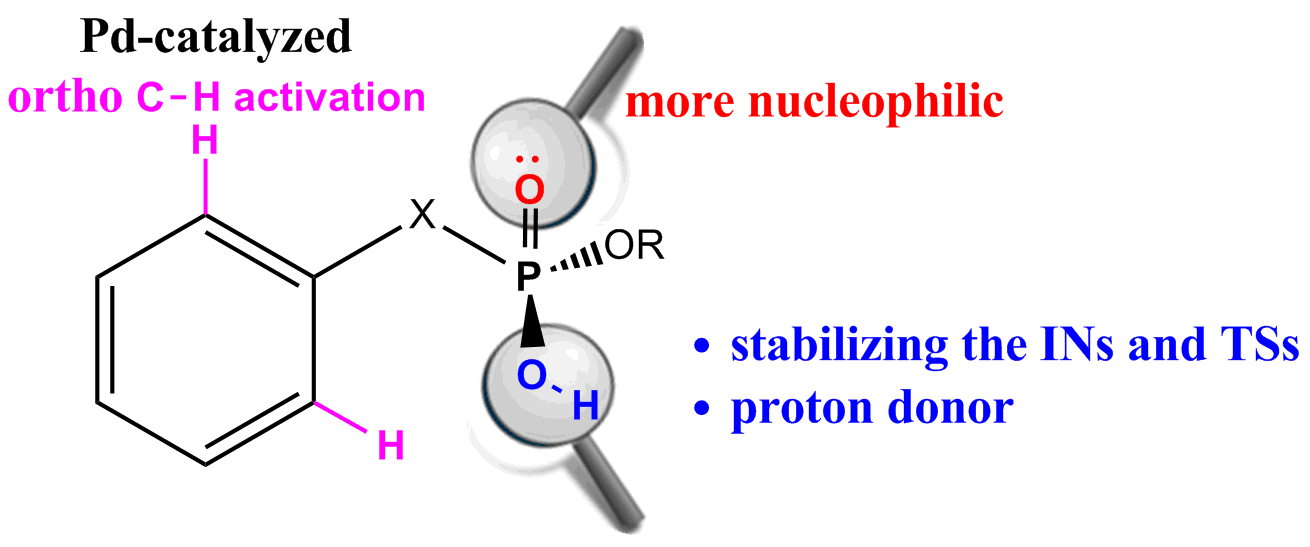
Probing Near-infrared Absorbance of E and Z Diazene Isomers via Antiaromaticity
Submitted by Jun Zhu on Tue, 08/15/2023 - 14:44
The photoswitching behaviors of heteroaryl azos and azobenzenes have attracted considerable interest due to their applications from material science to pharmacology. However, the use of UV light limits their application, especially in biomedicine and photopharmacology. In this work, using several aromaticity descriptors, including anisotropy of the induced current density analysis and nucleus-independent chemical shifts, we systematically investigate the relationship between anti-aromaticity and the absorption of a series of heterocyclic azos.