Mechanism, Reactivity, and Selectivity in Rh(III)-Catalyzed Phosphoryl-Directed Oxidative C–H Activation/Cyclization: A DFT Study
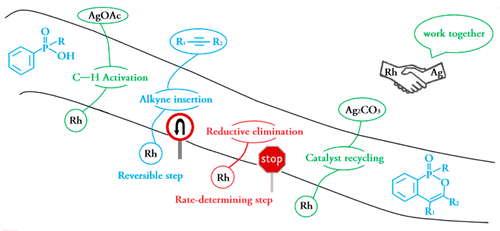
Density functional theory calculations (DFT) have been performed on Rh(III)-catalyzed phosphoryl-directed oxidative C–H activation/cyclization to investigate the detailed mechanism, including four basic steps: C–H activation, alkyne insertion, reductive elimination, and catalyst recycling, each of which consists of different steps. Interestingly, the Rh(III)–AgOAc catalyst system was found to be more favorable in the C–H activation step in comparison with the Rh(III)–Ag2CO3 system, whereas the Rh(I)–Ag2CO3 catalyst system was more efficient for catalyst recycling. Importantly, our calculations suggest that the alkyne insertion process is a reversible step. Reductive elimination is the rate-determining step with an activation energy of 25.0 kcal/mol. In addition, the origin of the reactivity and selectivity difference between diarylacetylenes and dialkylacetylenes or electron-rich and electron-deficient diarylacetylenes was probed by means of comparative DFT calculations. The calculation results show that the electronic effects of alkynes play a key role in the reactivity and selectivity, in line with the experimental observations that diarylacetylenes and electron-rich diarylacetylenes are more reactive than dialkylacetylenes and electron-deficient diarylacetylenes, respectively. Our findings should be useful for further developments of transition-metal-catalyzed C–H activation reactions.