Catalytic Mechanisms of Transfer Hydrogenation of Azobenzene with Ammonia Borane by Pincer Bismuth Complex: Crucial Role of C=N Functional Group on the Pincer Ligand
Submitted by Jun Zhu on Fri, 11/18/2022 - 22:13
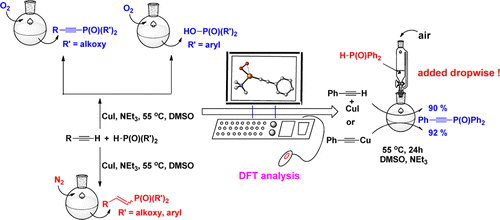
Transfer hydrogenation of azobenzene with ammonia borane mediated by pincer bismuth complex 1 were systematically investigated through density functional theory calculations. An unusual metal-ligand cooperation mechanism was disclosed, in which the saturation/regeneration of the C=N functional group on the pincer ligand plays an essential role. The reaction is initiated by the hydrogenation of the C=N bond (saturation) with ammonia borane to afford 3CN, which is the rate-determining step with Gibbs energy barrier (ΔG≠) and Gibbs reaction energy (ΔG) of 25.6 and -7.3 kcal/mol, respectively.