Substituent Modulated Adaptive Aromaticity in NHC-Pyrrolyl Cations: A Combined DFT and Machine Learning Study
Submitted by Jun Zhu on Mon, 05/25/2026 - 16:55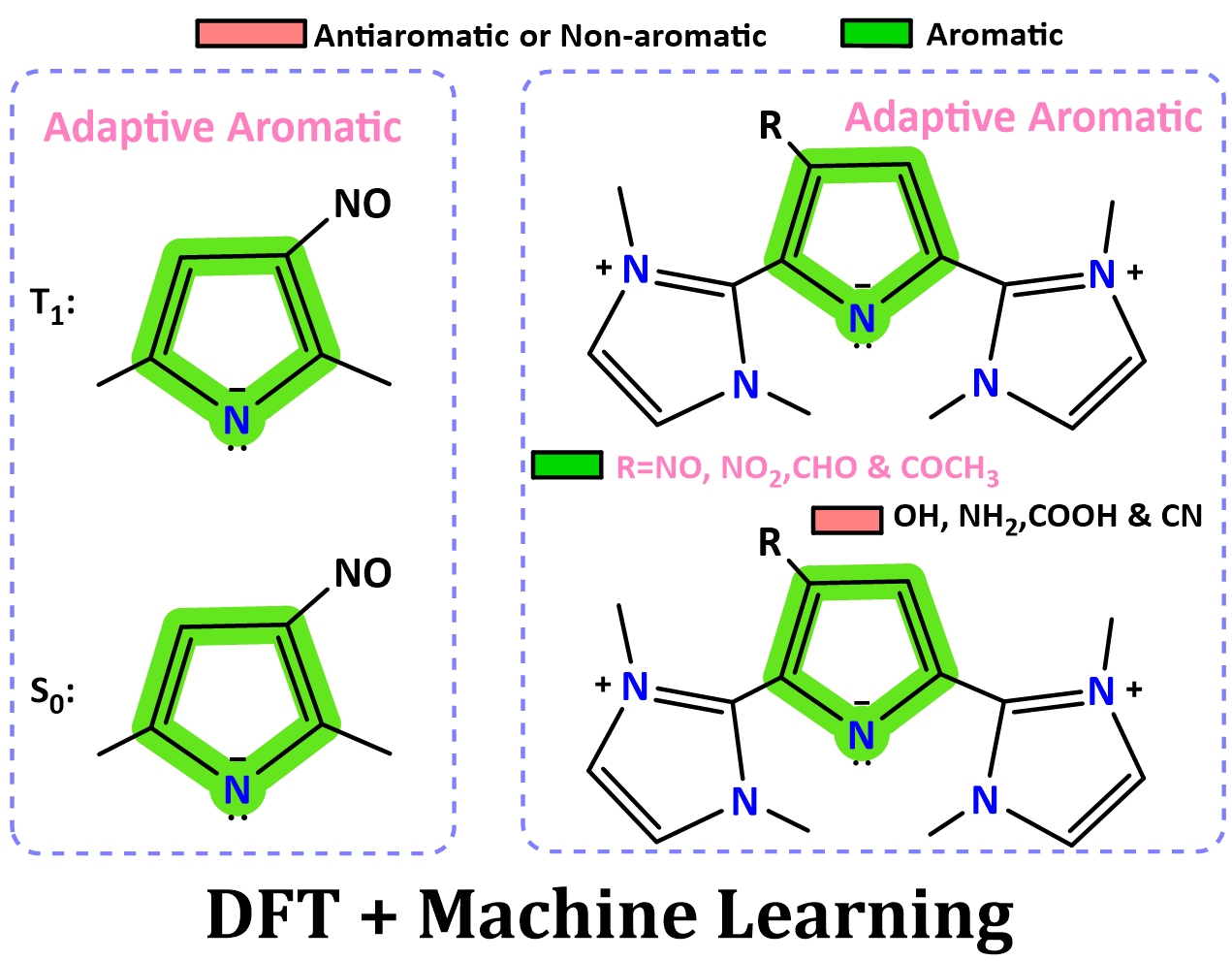
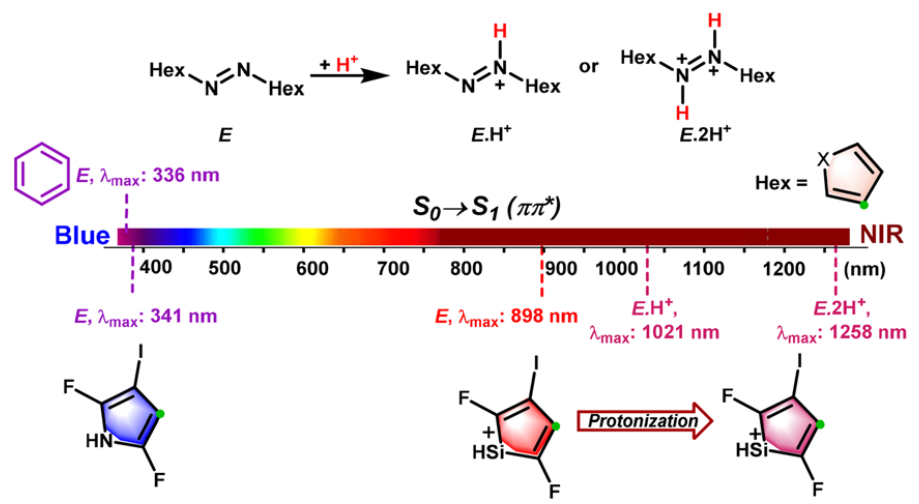
The stabilization of 4π-electron systems remains a fundamental challenge in chemistry, stemming from their intrinsic antiaromaticity and thermodynamic instability as dictated by Hückel’s rule. A recent experimental study has demonstrated that rational molecular design using N-heterocyclic carbenes (NHCs) can surmount these limitations by inducing aromaticity. However, the issue of T1 aromaticity in such systems remains unresolved. Elucidating the substituent-modulated behavior of T1 (anti)aromaticity is crucial for deciphering the physicochemical properties of these systems.