Static polarizabilities of copper cluster monocarbonyls CunCO (n=2-13) and selectivity of CO adsorption on copper clusters
Submitted by Jun Zhu on Thu, 10/31/2013 - 17:30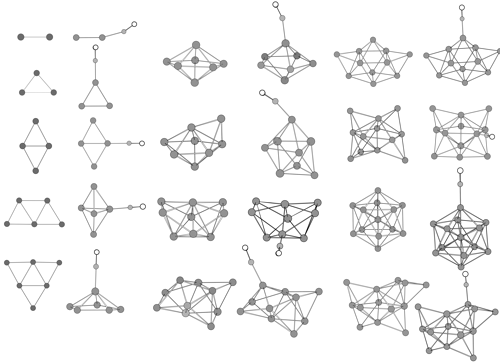
Density functional calculations for copper clusters Cu and their monocarbonyls CunCO (n less than or equal to 13) have been performed using the relativistic ECP plus DZ basis set augmented by an f polarization function for copper atom. Equilibrium geometries, harmonic frequencies, and static mean polarizabilities of Cu-n and CunCO are determined. The feature of CO adsorption on the copper cluster and the effect of CO adsorption on stability and polarizability of the cluster are investigated.
