Adaptive Aromaticity in 16-valence-electron Metallazapentalenes
Submitted by Jun Zhu on Tue, 11/09/2021 - 21:20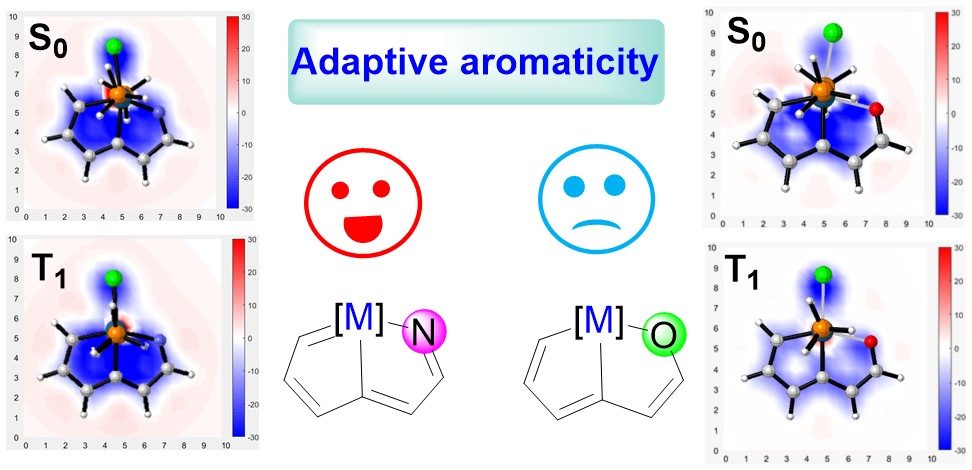
According to Hückel’s and Baird’s rules, cyclic species are generally aromatic only either in the lowest singlet state (S0) or in the lowest-lying triplet ππ* excited state (T1). Thus, species with aromaticity both in S0 and T1 states (termed as adaptive aromaticity) are particularly rare. Herein, we carry out density functional theory (DFT) calculations to examine the aromaticity of 16e metallapentalenes containing heteroatoms (N, O).