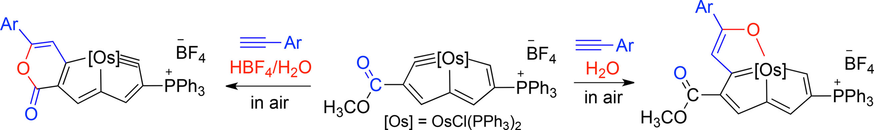
Utilization of a Chelating Bis[(dialkylamino)cyclopropenimine] to Isolate a Series of Heavier Zero-Valent Group 14 Tetracarbonyl Iron Complexes
Submitted by Jun Zhu on Tue, 04/22/2025 - 14:59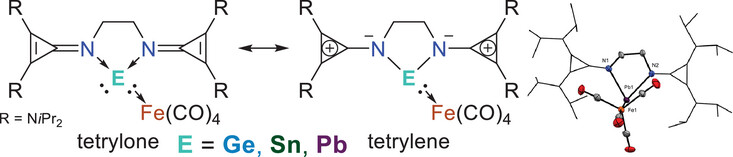
We report on the utilization of the ethylene-bridged bis[(dialkylamino)cyclopropenimine] (bisCPI) ligand, LCPI, to give access to new main-group E(II) halide complexes (E = Ge, Sn, Pb; 1, 2, 3). Subsequent reduction with Collman's reagent (Na2Fe(CO)4 • dioxane) enables the isolation of a series of zero-valent tetrylone-tetracarbonyl iron complexes, (LCPI)E(Fe(CO)4 (E = Ge (4), Sn (5), Pb (6)).