Screening Carbon-Boron Frustrated Lewis Pairs for Small-Molecule Activation including N2, O2, CO, CO2, CS2, H2O and CH4: A Computational Study
Submitted by Jun Zhu on Wed, 01/18/2023 - 22:36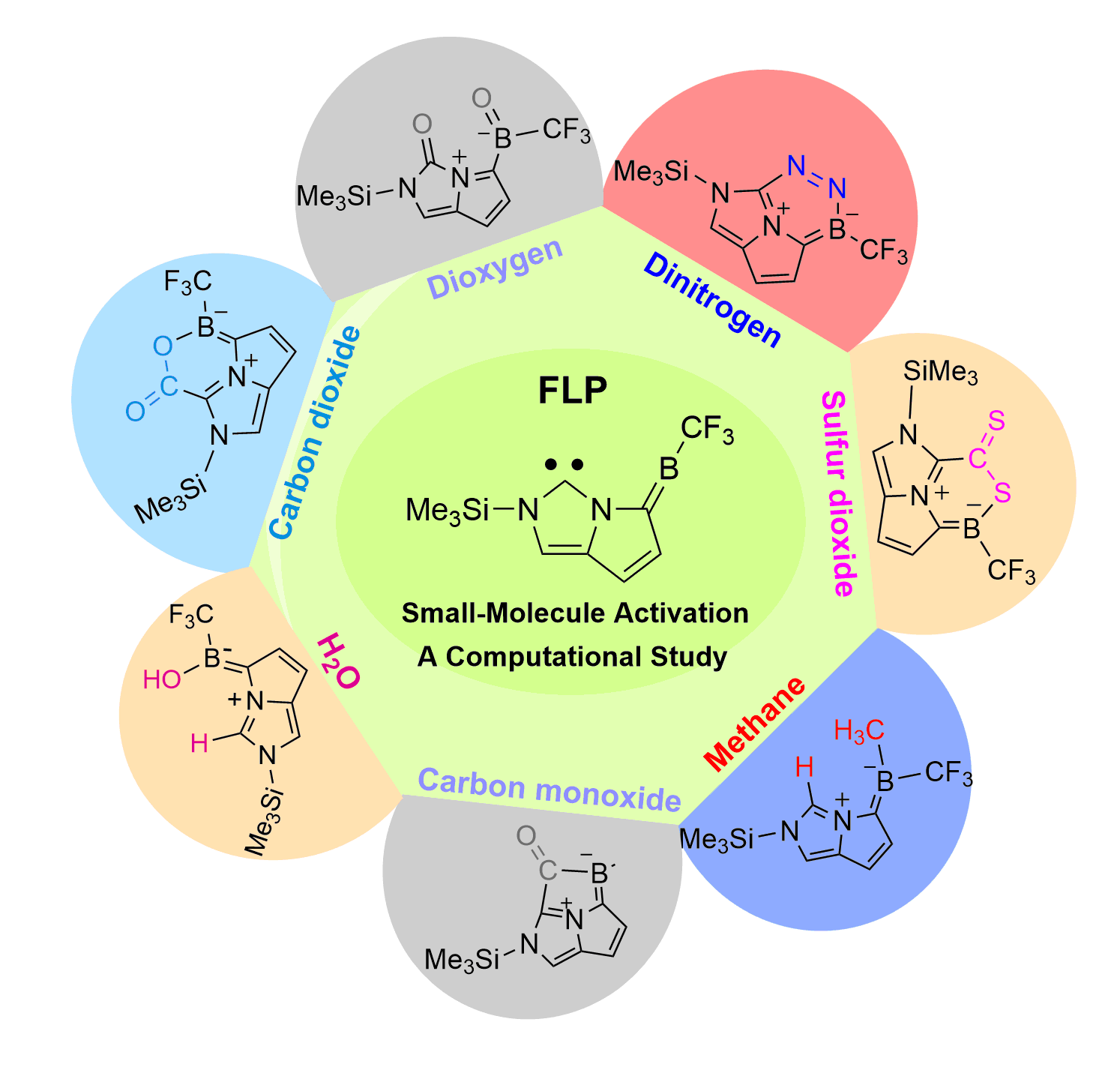
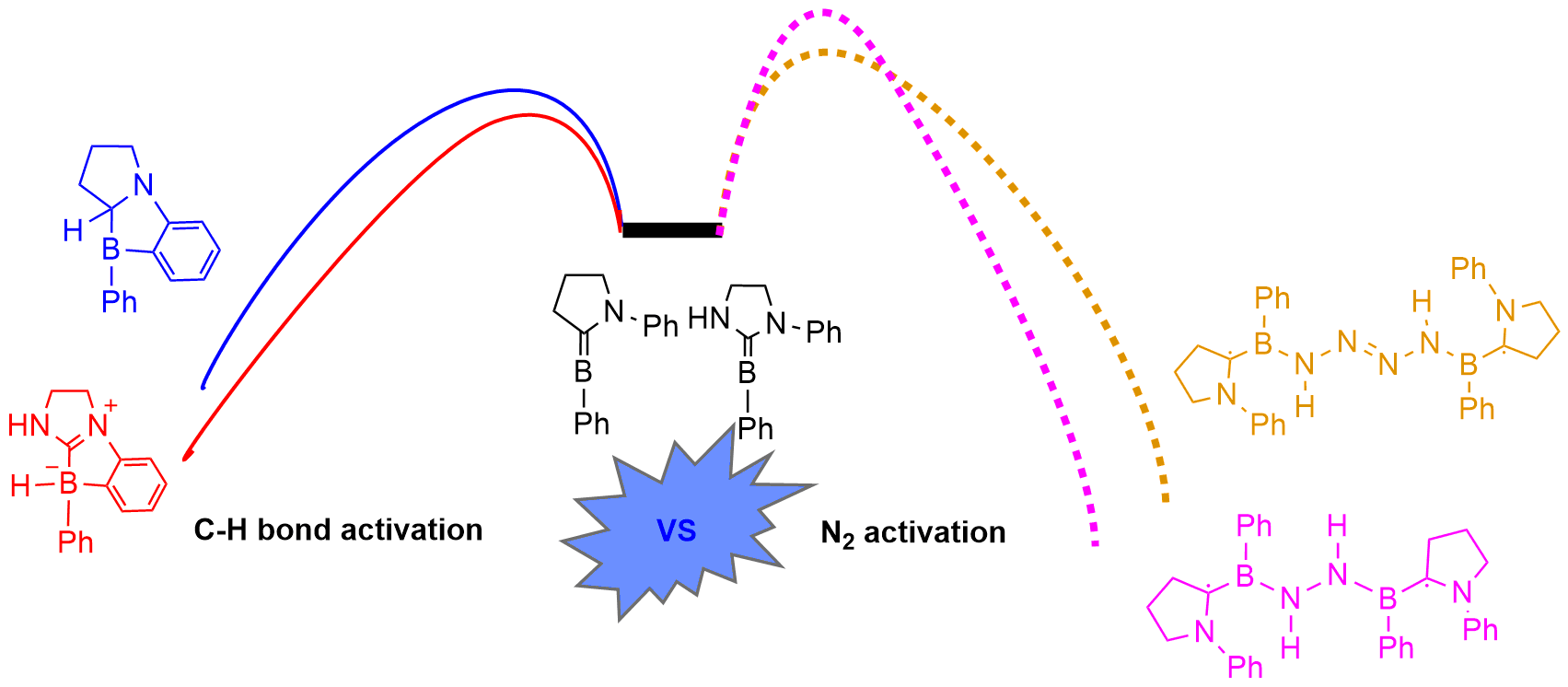
Dinitrogen (N2) activation is particularly challenging under ambient conditions because of its large highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) gap (10.8 eV) and high bond dissociation energy (945 kJ mol–1) of the NΞN triple bond, attracting considerable attention from both experimental and theoretical chemists. However, most effort has focused on metallic systems. In contrast, nitrogen activation by frustrated Lewis pairs (FLPs) has been initiated recently via theoretical calculations.