Theoretical Study on Reaction Mechanisms of Dinitrogen Activation and Coupling by Carbene-Stabilized Borylenes in Comparison with Intramolecular C-H Bond Activation
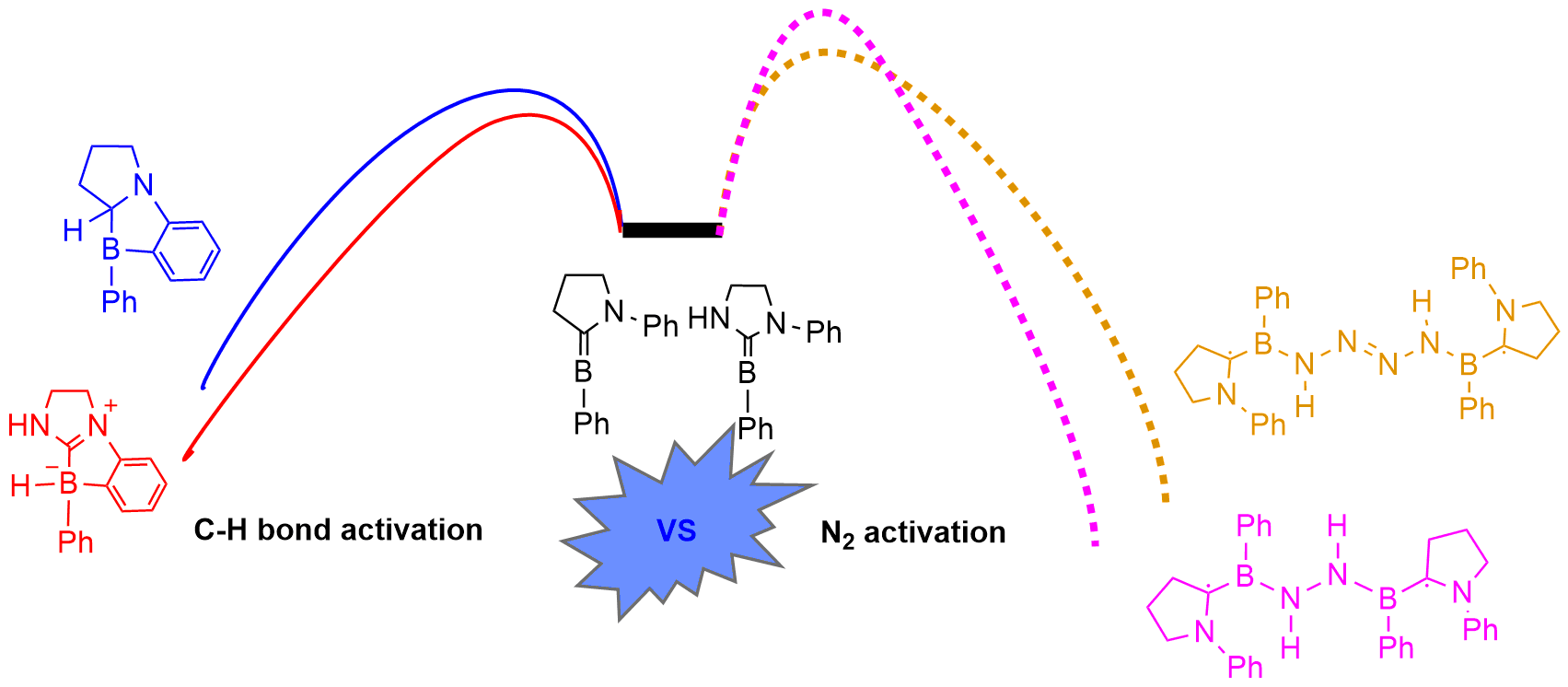
Dinitrogen (N2) activation is particularly challenging due to the significantly strong N≡N bond, let alone the catenation of two N2 molecules. Recent experimental study shows that cyclic (alkyl)(amino)carbene (CAAC)-stabilized borylenes are able to tackle N2 activation and coupling below room temperature. Here we carry out density functional theory calculations to explore the corresponding reaction mechanisms. The results indicate that the reaction barrier for the dinitrogen activation by the first borylene is slightly higher than that by the second borylene. In addition, replacing the CAAC moiety of the borylenes with cyclic diaminocarbenes (CDACs) could make such dinitrogen activation and coupling more favorable thermodynamically. The reaction mechanisms of the intramolecular C-H bond activation of borylene have also been discussed, which is found to be favorable both thermodynamically and kinetically in comparison with N 2 activation. Thus, adequate attention should be paid to the design of borylenes aiming at N2 activation. In addition, our calculations suggest that the CDAC moiety of the borylene could lead to a different product in terms of intramolecular C-H bond activation. All these findings could be useful for the development of dinitrogen activation as well as C-H bond activation by main group species.