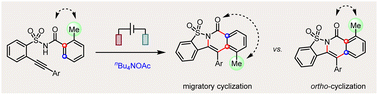
Direct amination of anilines utilizing dearomatized phenolate species
Submitted by Jun Zhu on Wed, 05/21/2025 - 15:20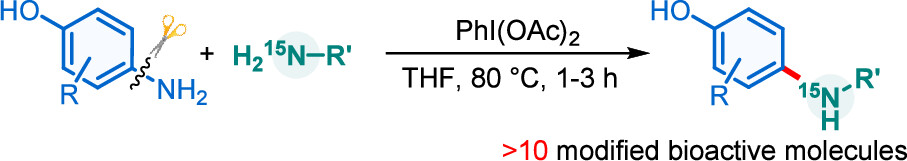
Activation of the aryl C–N bond underpins critical challenges in modern organic synthesis. Herein, the direct amination of anilines is presented via hypervalent iodine-mediated transient dearomatized phenolate intermediates, enabling selective C(aryl)–NH2 bond cleavage under mild conditions. A library of bioactive p-alkylaminophenols is synthesized in up to 85% yields within 3 h. Being used in late-stage drug diversification and mechanistic studies, this protocol offers a modular platform for complex amine construction.