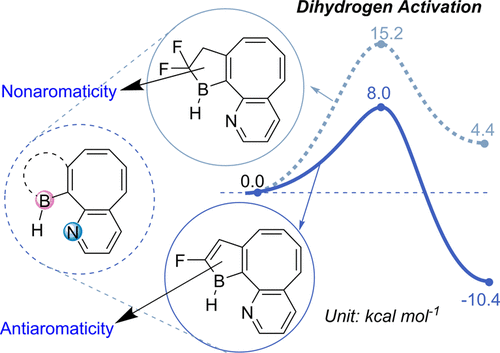
Carbon-halogen bond activation by a structurally constrained phosphorus(III) platform
Submitted by Jun Zhu on Mon, 11/09/2020 - 11:27
The σ-bond activation by main group element has received enormous attention from theoretical and experimental chemists. Here, the reaction of C-X (X = Cl, Br, I) bonds in benzyl and allyl halides with a pincer-type phosphorus(III) species was reported. A series of structurally robust phosphorus(V) compounds were formed via the formal oxidative addition reactions of C-X bonds to the phosphorus(III) center. Density functional theory calculations show that the nucleophilic addition process is more favorable than the direct oxidative addition mechanism.