Mechanism, catalysis and predictions of 1,3,2-diazaphospholenes: theoretical insight into highly polarized P–X bonds
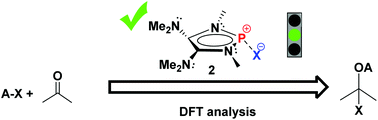
Density functional theory (DFT) calculations were carried out to investigate the hydridic character of several main group hydrides. A P-hydrido-1,3,2-diazaphospholene 1f with two π-electron donor amino groups on the heterocyclic skeleton framework performs as a strong hydride donor owing to the significant n(N)–σ*(P–H) hyperconjugation. The natural bond orbital analysis reveals that high π-electron delocalization exists in both 1f and the corresponding stable phosphenium Ef+. In addition, 1f is calculated to have a similar catalytic ability for the hydroboration of acetone with pinacolborane, compared to 1e. Thus, a variety of organic substrates activated by 1f are explored, including ketone, imine, isocyanate, CO2, diazene, alkene, alkyne and epoxide. The results show that the highly polarized and electron-deficient bonds such as CO π bonds are readily activated, whereas 1f seems difficult to react with electron-rich unsaturated bonds of propene and propyne. More importantly, 1,3,2-diazaphospholene-based compounds, featuring an extremely polarized P–X bond (X = CCMe, NMe2, PMe2 and SMe), are predicted to have a useful catalytic ability. The preliminary computational results suggest that these P–X compounds could catalyze the silylamination, silylphosphination and silylsulfenylation of acetone with TMSNMe2, TMSPMe2 and TMSSMe, respectively. The products are silylethers, which are equivalent to the corresponding alcohols since they easily undergo hydrolysis. Our computational study opens a new avenue to the design of novel main group organocatalysts.
http://pubs.rsc.org/en/content/articlelanding/2016/qo/c6qo00002a#!divAbstract