Single-Molecule Resolved Conformational and Orbital Symmetry Breaking in Tetraphenylethylene-Based Macrocycles
Submitted by Jun Zhu on Sun, 12/08/2024 - 10:38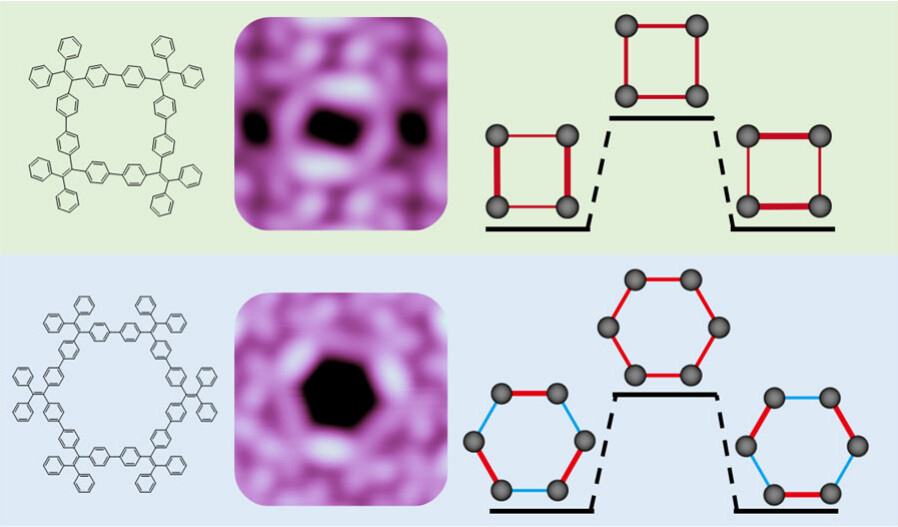
Tetraphenylethylene (TPE) is a prototype aggregate-induced emission molecule. TPE-based conjugated macrocycles exhibit unique optical properties due to their peculiar cyclic topology. Because the symmetry of macrocycles strongly affects their photophysical properties, here we report a single-molecule study of the structures and orbitals of two TPE-based macrocycles of (C26H18)4 and (C26H18)6. Using scanning tunneling microscopy and spectroscopy, we discover that both macrocycles undergo spontaneous symmetry breaking in their conformations and frontier orbitals.