Probing the origin of the stereoselectivity and enantioselectivity of cobalt-catalyzed [2 + 2] cyclization of ethylene and enynes
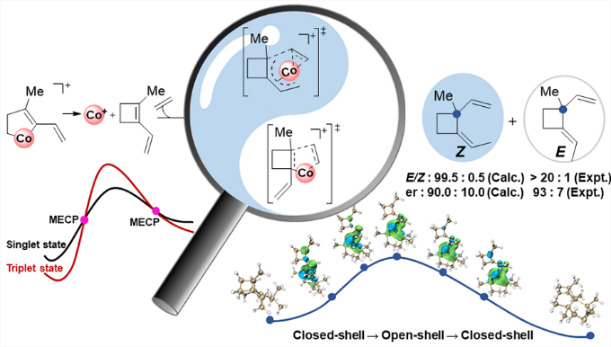
The cyclobutane unit is important to prepare complex natural products with biological activity due to the high ring strain. Among various approaches, [2 + 2] cycloaddition is one of the major strategies to prepare cyclobutane under light conditions. Recently, Rajanbabu's group reported tandem catalysis for asymmetric coupling of inactivated ethylene and enynes to functionalized cyclobutenes or cyclobutanes. However, the reaction mechanisms remain unproven. Herein, we demonstrate via comprehensive density functional theory calculations that the computed reaction mechanism for the formation of cyclobutene is in line with the originally proposed mechanism. However, the proposed mechanism for the formation of cyclobutane is computed to be unfavorable both thermodynamically and kinetically. Specifically, calculations reveal that a direct β-hydrogen transfer is more favorable than β-hydrogen elimination in the formation of (Z)-cyclobutane. More interestingly, several minimum energy crossing points (MECPs) are located along various potential energy surfaces, highlighting the importance of spin-crossing. All these findings could be particularly useful for the development of asymmetric Co-catalyzed [2 + 2] cyclization.
https://pubs.rsc.org/en/content/articlelanding/2021/QO/D0QO01412E#!divAbstract