Understanding nonplanarity in metallabenzene complexes
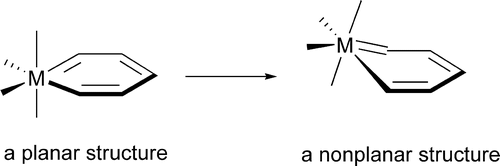
The nonplanarity found in metallabenzene complexes has been investigated theoretically via density functional theory (DFT) calculations. A metallabenzene has four occupied π molecular orbitals (8 π electrons) instead of three that benzene has. Our electronic structure analyses show that the extra occupied π molecular orbital, which is the highest occupied molecular orbital (HOMO) in many metallabenzenes, has antibonding interactions between the metal center and the metal-bonded ring-carbon atoms, providing the electronic driving force toward nonplanarity. Calculations indicate that the electronic driving force toward nonplanarity, however, is relatively small. Therefore, other factors such as steric effects also play important roles in determining the planarity of these metallabenzene complexes. In this paper, how the various electronic and steric factors interplay has been discussed.