Understanding nonplanarity in metallabenzene complexes
Submitted by Jun Zhu on Thu, 10/31/2013 - 23:54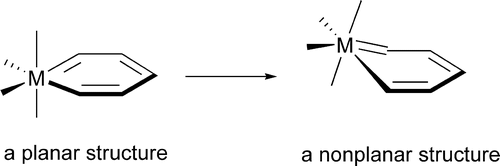
The nonplanarity found in metallabenzene complexes has been investigated theoretically via density functional theory (DFT) calculations. A metallabenzene has four occupied π molecular orbitals (8 π electrons) instead of three that benzene has. Our electronic structure analyses show that the extra occupied π molecular orbital, which is the highest occupied molecular orbital (HOMO) in many metallabenzenes, has antibonding interactions between the metal center and the metal-bonded ring-carbon atoms, providing the electronic driving force toward nonplanarity.