Interpolation of Atomically Thin Hexagonal Boron Nitride and Graphene: Electronic Structure and Thermodynamic Stability in Terms of All-Carbon Conjugated Paths and Aromatic Hexagons
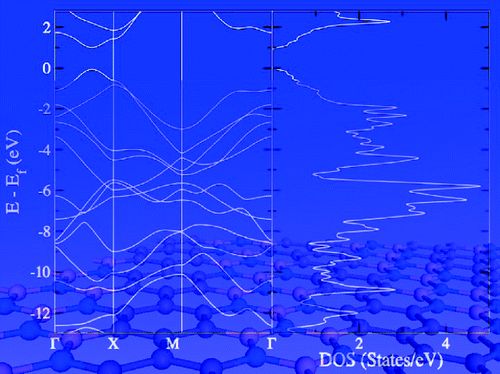
Two-dimensional hexagonal composite materials (BN)n(C2)m (n, m = 1, 2, ...), which all are isoelectronic with graphene and hexagonal boron nitride (h-BN), have been studied by density functional theory (DFT) with a focus on the relative energies of different material isomers and their band gaps. The well-established chemical concepts of conjugation and aromaticity were exploited to deduce a rationale for identifying the thermodynamically most stable isomer of the specific composites studied. We find that (BN)n(C2)m materials will not adopt structures in which the B, C, and N atoms are finely dispersed in the 2D sheet. Instead, the C atoms and C−C bonds, which provide for improved conjugation when compared to B−N bonds, gather and form all-carbon hexagons and paths; that is, the (BN)n(C2)m materials prefer nanostructured distributions. Importantly, there are several isomers of similarly low relative energy for each (BN)n(C2)m composite type, but the band gaps for these nearly isoenergetic isomers differ by up to 1.0 eV. This feature in the band gap variation of the most stable few isomers is found for each of the four composites studied and at two different DFT levels. Consequently, the formation of a distinct (BN)n(C2)m material isomer with a precise (small) band gap will likely be nontrivial. Therefore, one likely has to invoke nonstandard preparation techniques that exploit nanopatterned h-BN or graphene with voids that can be filled with the complementary all-carbon or boron nitride segments.