Unprecedented bis(silylene)-supported silylone–metal complexes with Si0→CuI, Si0→NiI, and Si0→NiII dative bonds
Submitted by Jun Zhu on Wed, 05/21/2025 - 15:38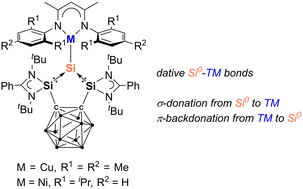
The first silylone-3d-metal complexes, LSiCu(NacNacM) (2) [L = 1,2-(RSi)2-1,2-C2B10H10, R = PhC(NtBu)2; NacNacM = HC(CMeNMes)2, Mes = 2,4,6-Me3-C6H2] and LSiNi(NacNacD) (3) [NacNacD = HC(CMeNDipp)2, Dipp = 2,6-iPr2-C6H3], are reported, resulting from the reaction of the strongly σ-donating and chelating bis(silylenyl)-ortho-carborane silylone LSi0 with [(NacNacMCu)2benzene] and [(NacNacDNi)2toluene], respectively.