Switchable Reactivities of Metalated Phosphasilenes Regulated by Reversible 1,2-Metal Migration
Submitted by Jun Zhu on Tue, 01/20/2026 - 10:40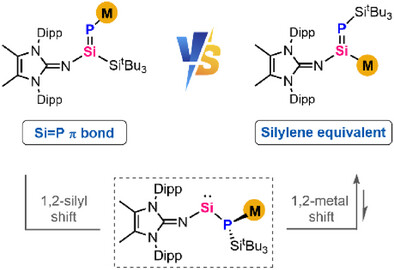
Anionic reagents with silicon-containing double bonds, M(R)Si═ERn (E = main group elements), have garnered significant interest owing to their unique metal-mediated reactivity and their potential in transferring the Si═E unit. Within this domain, the intriguing field of Si-metalated phosphasilenes remains uncharted.