β-Hydrogen Elimination of Five-Membered-Ring Metallacycles. Is It Possible?
Submitted by Jun Zhu on Thu, 10/31/2013 - 22:53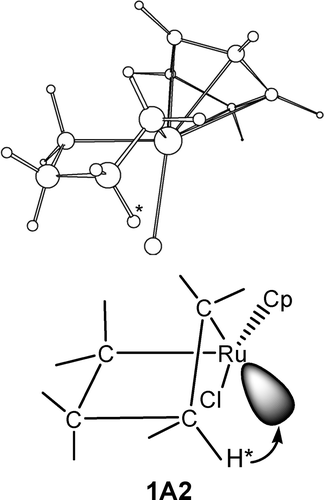
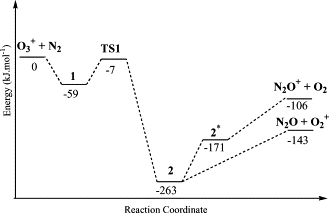
B3LYP density functional theory calculations have been carried out to examine the structural and energetic aspects of β-hydrogen elimination in several metallacyclic complexes of ruthenium and platinum. Factors affecting barriers of the elimination reactions have been examined. It was found that favorable structural arrangements, in which the transferring β-hydrogen is in close proximity to the metal center, for β-hydrogen elimination exist in certain ring conformations of metallacyclic complexes.