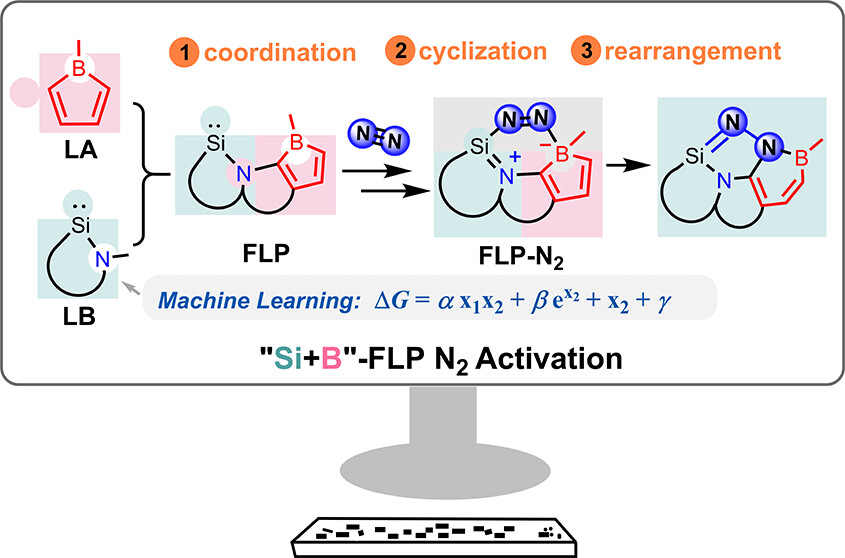
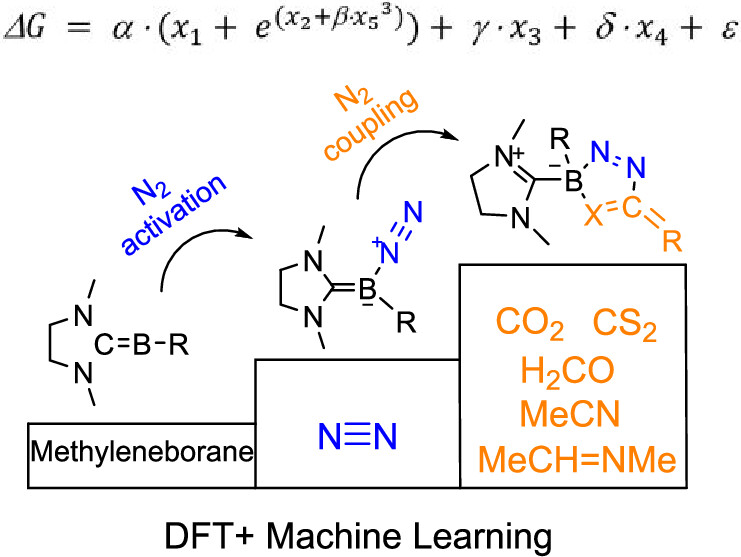
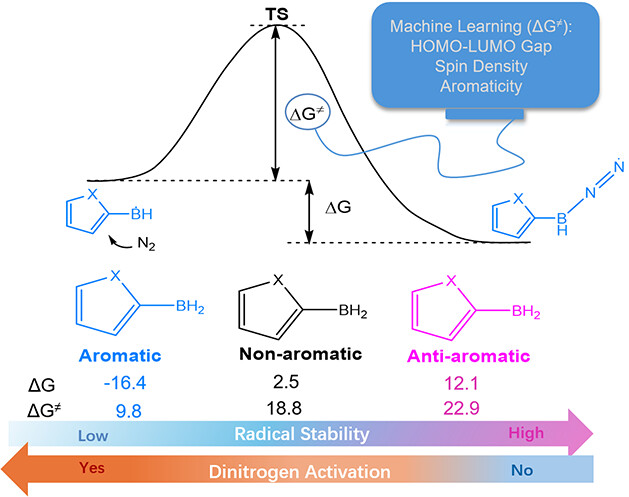
Charge-Promoted Adaptive Aromaticity in Metallabenzenes: A Combined DFT and Machine Learning Study
Submitted by Jun Zhu on Mon, 01/26/2026 - 16:15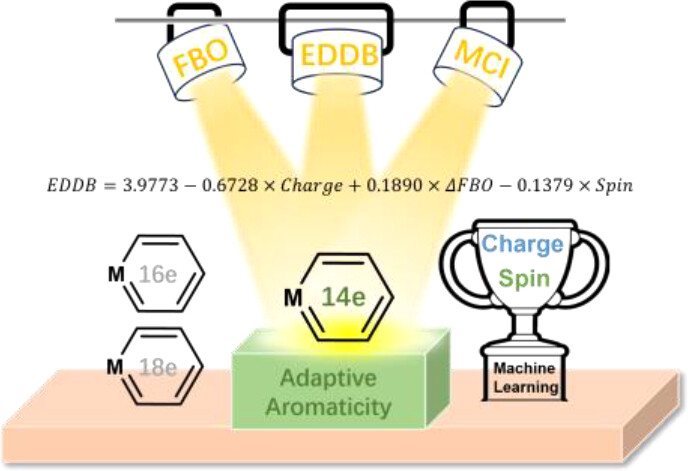
In accordance with the constraints by Hückel’s and Baird’s rules, species generally exhibit aromaticity in one state (the lowest singlet state S0 or the lowest triplet state T1). Consequently, species with adaptive aromaticity (being aromatic in both the S0 and T1 states) are particularly rare. In this study, density functional theory (DFT) was employed to investigate adaptive aromaticity in 14e–, 16e– and 18e– metallabenzenes.