Aromaticity Effects on the Profiles of the Lowest Triplet-State Potential-Energy Surfaces for Rotation about the CC Bonds of Olefins with Five-Membered Ring Substituents: An Example of the Impact of Baird's Rule
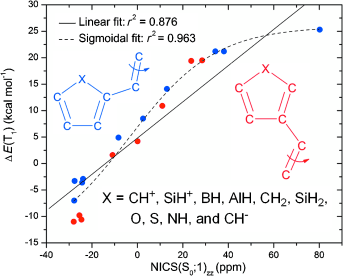
A density functional theory study on olefins with five-membered monocyclic 4n and 4n+2 π-electron substituents (C4H3X; X=CH+, SiH+, BH, AlH, CH2, SiH2, O, S, NH, and CH−) was performed to assess the connection between the degree of substituent (anti)aromaticity and the profile of the lowest triplet-state (T1) potential-energy surface (PES) for twisting about olefinic CC bonds. It exploited both Hückel’s rule on aromaticity in the closed-shell singlet ground state (S0) and Baird’s rule on aromaticity in the lowest ππ* excited triplet state. The compounds CH2CH(C4H3X) were categorized as set A and set B olefins depending on which carbon atom (C2 or C3) of the C4H3X ring is bonded to the olefin. The degree of substituent (anti)aromaticity goes from strongly S0-antiaromatic/T1-aromatic (C5H4+) to strongly S0-aromatic/T1- antiaromatic (C5H4−). Our hypothesis is that the shapes of the T1 PESs, as given by the energy differences between planar and perpendicularly twisted olefin structures in T1 [ΔE(T1)], smoothly follow the changes in substituent (anti)aromaticity. Indeed, correlations between ΔE(T1) and the (anti)aromaticity changes of the C4H3X groups, as measured by the zz-tensor component of the nucleus-independent chemical shift ΔNICS(T1;1)zz, are found both for sets A and B separately (linear fits; r2=0.949 and 0.851, respectively) and for the two sets combined (linear fit; r2=0.851). For sets A and B combined, strong correlations are also found between ΔE(T1) and the degree of S0 (anti)aromaticity as determined by NICS(S0,1)zz (sigmoidal fit; r2=0.963), as well as between the T1 energies of the planar olefins and NICS(S0,1)zz (linear fit; r2=0.939). Thus, careful tuning of substituent (anti)aromaticity allows for design of small olefins with T1 PESs suitable for adiabatic Z/E photoisomerization.