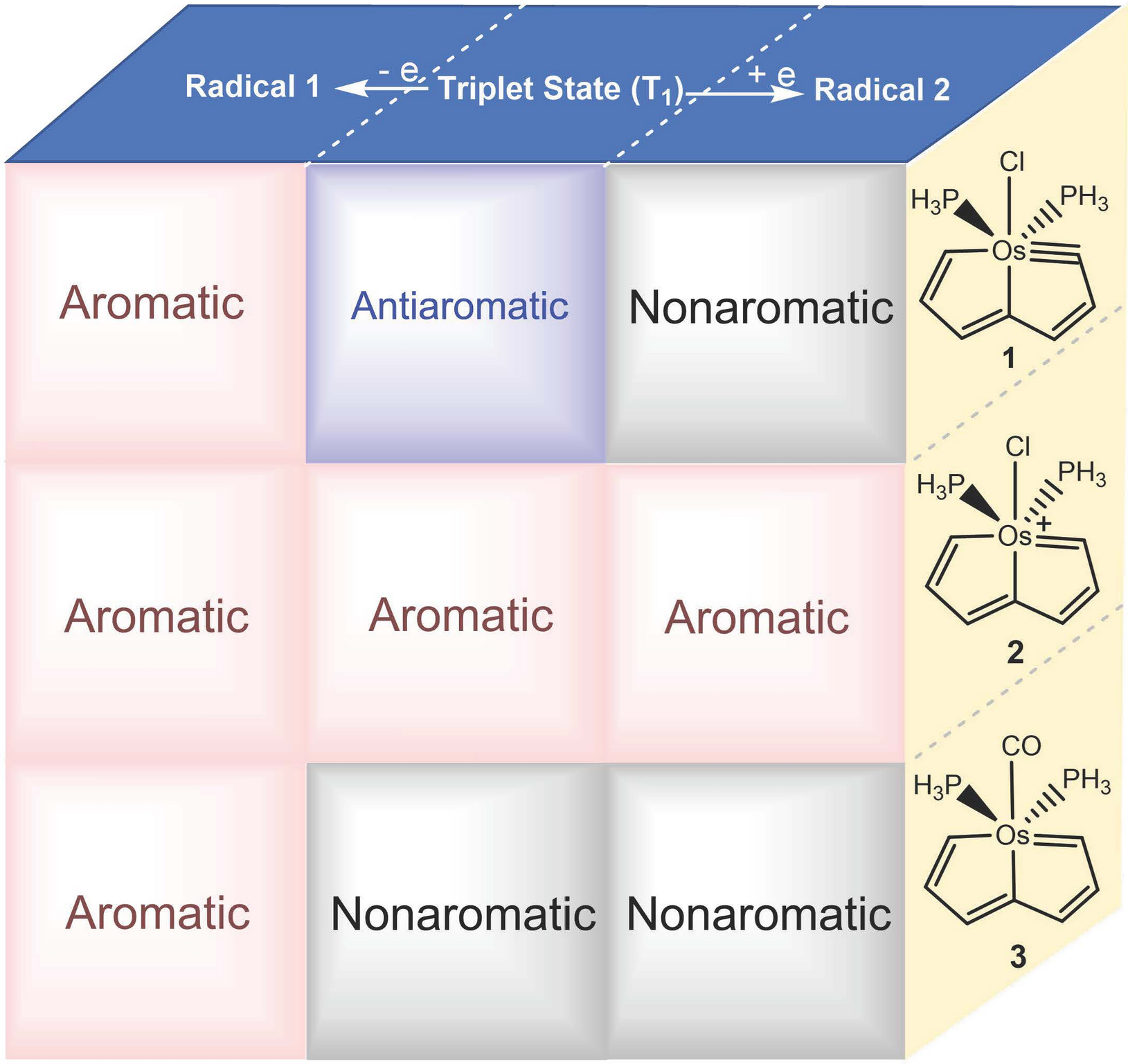
Formally Triplet Aluminyl Anions within the [Al2Pd2]2– Cluster Stabilized by All-Metal Double Aromaticity
Submitted by Jun Zhu on Mon, 03/16/2026 - 09:52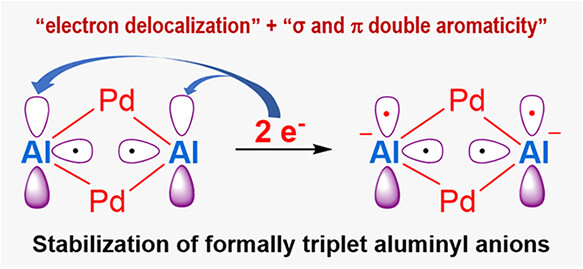
While triplet carbenes have been stabilized through spin delocalization, capturing triplet aluminyl anions─isoelectronic counterparts of carbenes─has remained an unsolved challenge in main-group chemistry. All previously reported aluminyl anions exhibit a singlet ground state, despite the use of diverse ligand frameworks. Herein, we report the first example of formally triplet aluminyl anions within bimetallic clusters, [((CH3)2C(CH2NPiPr2)2)2Al2Pd2]M2 (M = Li, Na, K).