Phosphine-Stabilized Germylidenylpnictinidenes as Synthetic Equivalents of Heavier Nitrile and Isocyanide in Cycloaddition Reactions with Alkynes
Submitted by Jun Zhu on Fri, 03/11/2022 - 07:53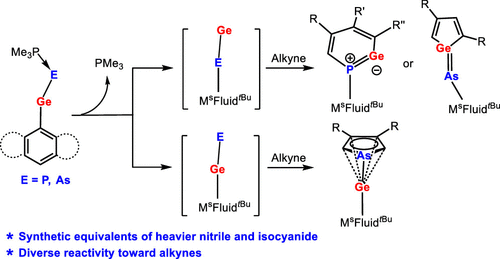
The reactions of chlorogermylene MsFluindtBu-GeCl 1, supported by a sterically encumbered hydrindacene ligand MsFluindtBu, with NaPCO(dioxane)2.5 and NaAsCO(18-c-6) in the presence of trimethylphosphine afforded trimethylphosphine-stabilized germylidenyl-phosphinidene 2 and -arsinidene 3, respectively. Structural and computational investigations reveal that the Ge–E′ bond (E′ = P and As) features a multiple-bond character.