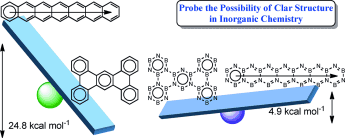
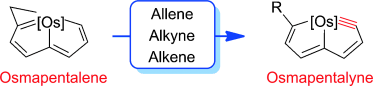
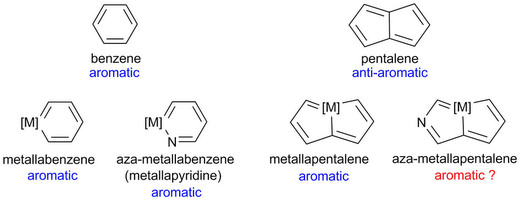
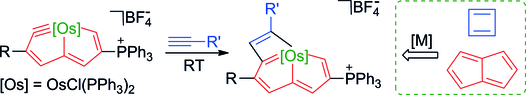
Synthesis of tetra- and octa-aurated heteroaryl complexes towards probing aromatic indoliums
Submitted by Jun Zhu on Mon, 05/23/2016 - 20:01
Polymetalated aromatic compounds are particularly challenging synthetic goals because of the limited thermodynamic stability of polyanionic species arising from strong electrostatic repulsion between adjacent carbanionic sites. Here we describe a facile synthesis of two polyaurated complexes including a tetra-aurated indole and an octa-aurated benzodipyrrole. The imido trinuclear gold(I) moiety exhibits nucleophilicity and undergoes an intramolecular attack on a gold(I)-activated ethynyl to generate polyanionic heteroaryl species.