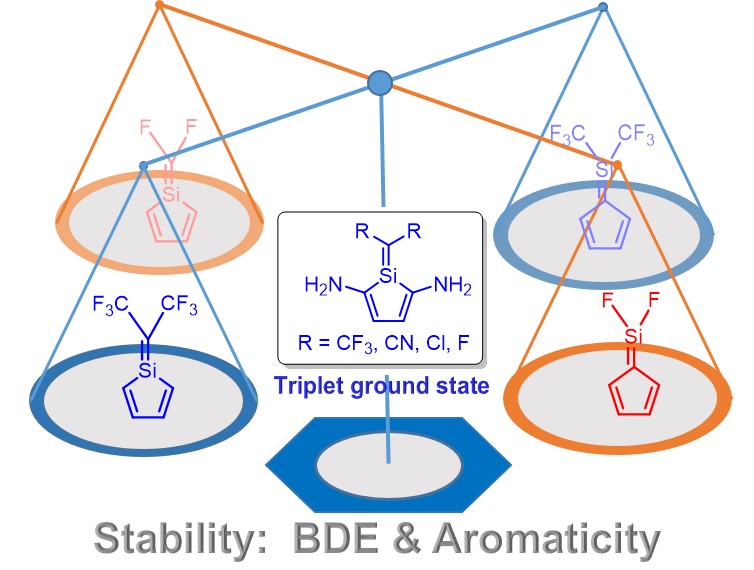
Probing the Aromaticity and Stability of Metallatricycles by DFT Calculations: Toward Clar Structure in Organometallic Chemistry
Submitted by Jun Zhu on Fri, 02/28/2020 - 15:49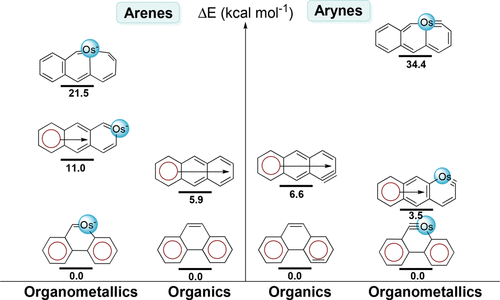
Metallaaromatics have attracted considerable attention in recent years because they can display properties of both organic and organometallic species. However, it remains unclear whether Clar’s rule could be applied to organometallic chemistry despite its proposal in 1950s. Here, we investigate the relative stabilities of 49 organic and organometallic species by density functional theory (DFT) calculations.