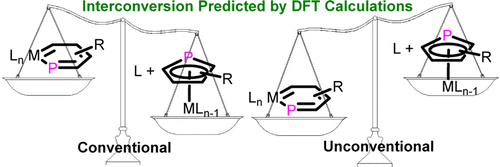
Unconventional Facile Way to Metallanaphthalenes from Metal Indenyl Complexes Predicted by DFT Calculations: Origin of Their Different Thermodynamics and Tuning Their Kinetics by Substituents
Submitted by Jun Zhu on Thu, 04/24/2014 - 22:47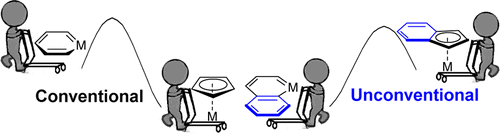
Metallaaromatics have attracted considerable interest from both experimentalists and theoreticians over the past three decades. However, most studies in this field have focused on metallabenzene, in which a CH group is replaced by a transition metal fragment. In comparison with monocyclic metallabenzenes, bicyclic metallanaphthalenes are rather limited. Thus, it is urgent to explore more synthetic approaches to this less developed system. One of the difficulties in the synthesis of metallanaphthalenes could be due to its low thermodynamic stability relative to the metal indenyl complexes.