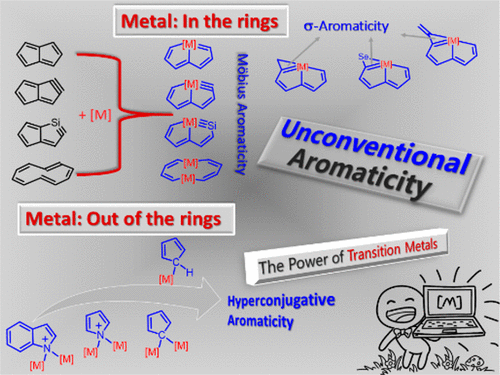
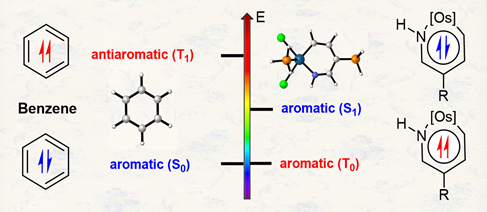
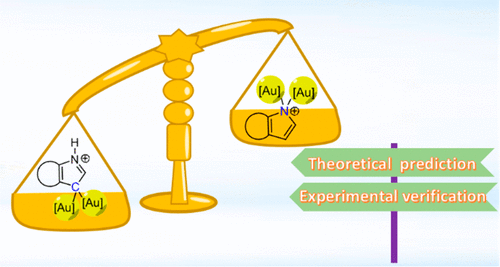
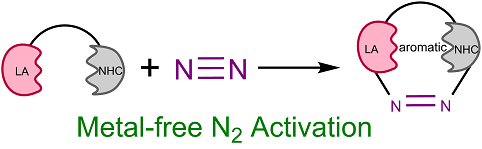
3‐Aminoindole Synthesis from 2‐Nitrochalcones and Ammonia or Primary Amines
Submitted by Jun Zhu on Wed, 07/03/2019 - 21:26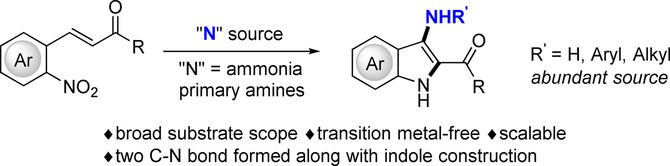
A step‐economic strategy for 3‐aminoindoles synthesis with ammonia or primary amines as “N” source under transition‐metal‐free conditions was achieved. A series of 3‐aminoindoles was obtained with abundant “N” source featuring high efficiency, mild conditions, environmental friendliness and scalability. Efficient syntheses of the intermediates of COX‐2 inhibitor and tubulin polymerization inhibitor were successfully accomplished with this newly developed strategy.