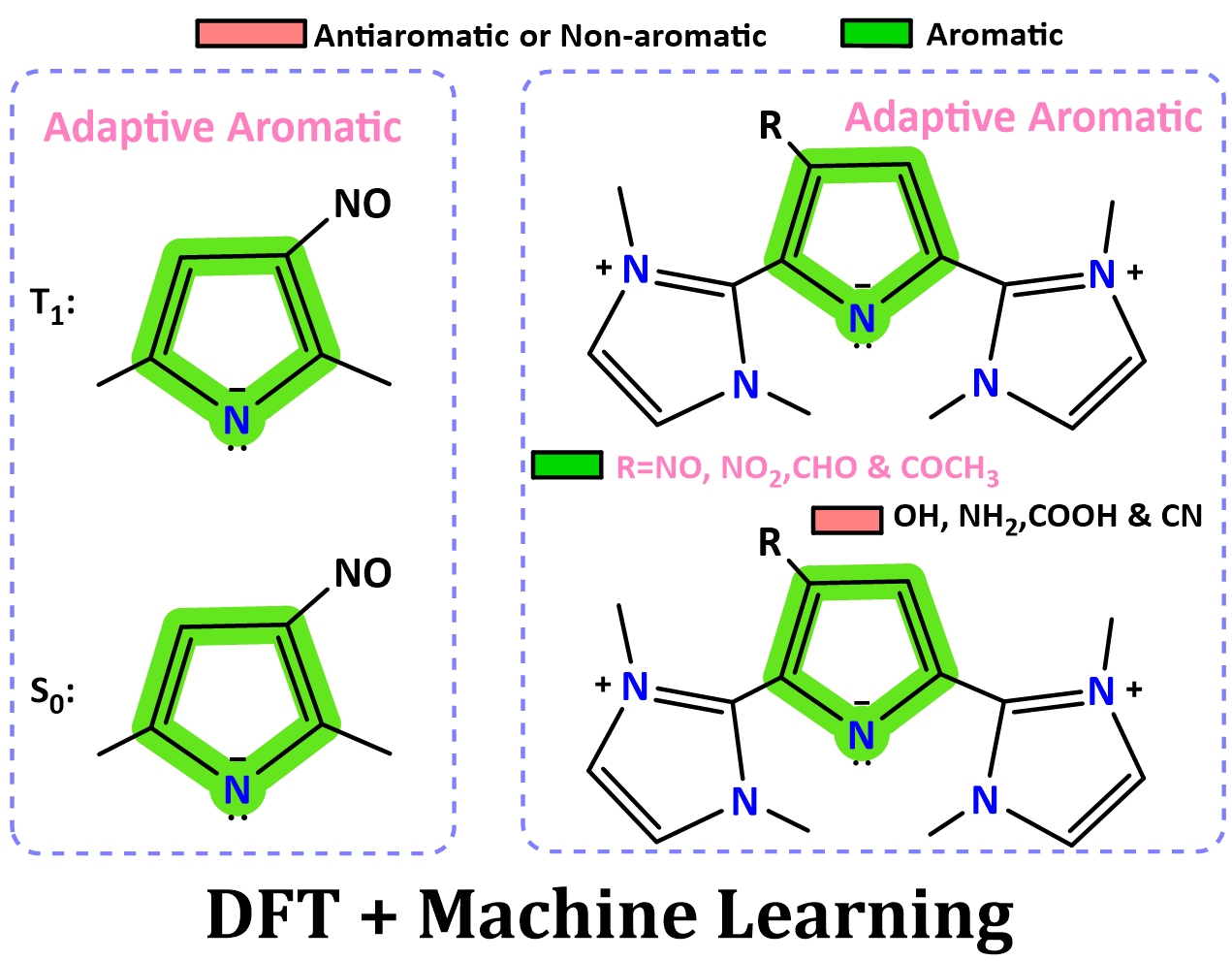
Electron-injection-induced global aromaticity enables stable open-shell nanopillars with intense mid-infrared magnetic circular dichroism
Submitted by Jun Zhu on Sat, 06/06/2026 - 09:02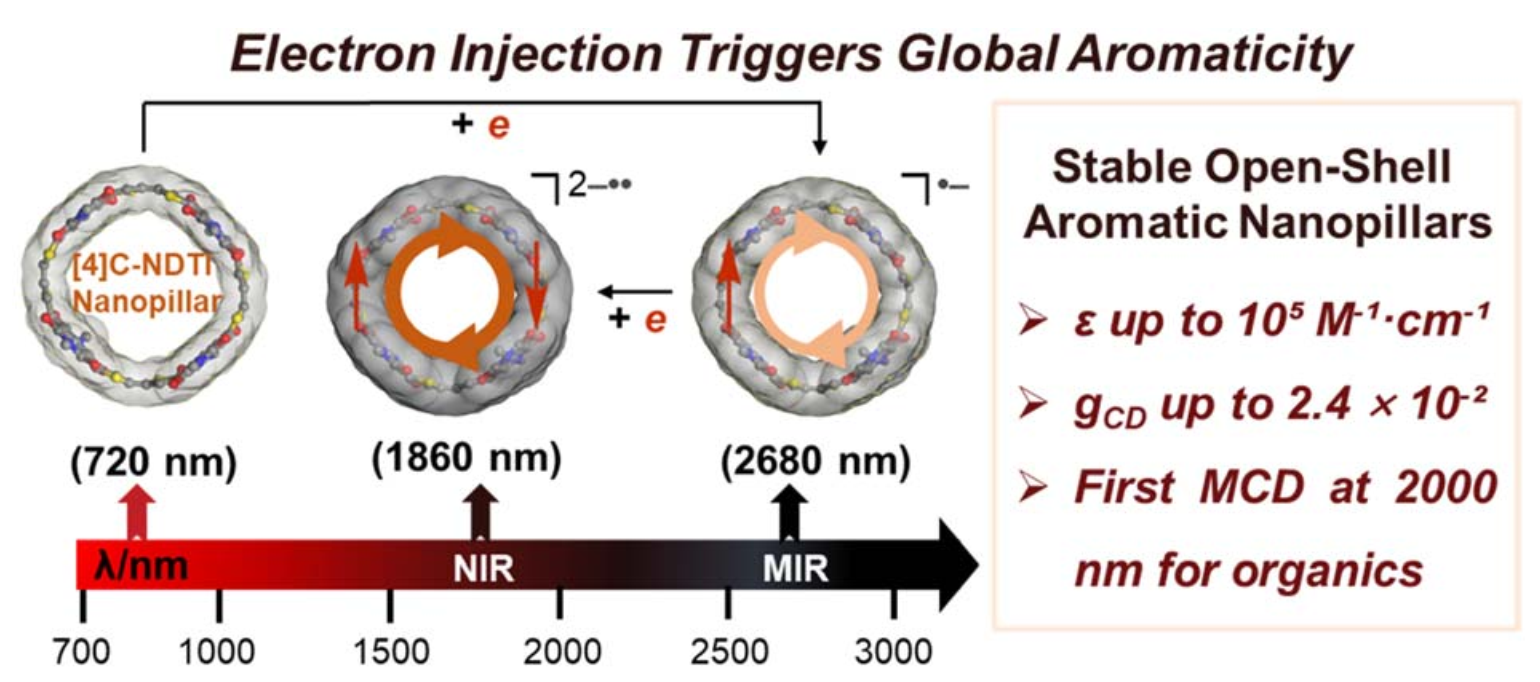
Open-shell species are central to spintronics and infrared optoelectronics, but remain challenging to stabilize in discrete molecular systems. Herein, we report that electron injection into pillar-shaped, radially π-conjugated [4]cyclonaphthodithiophene diimides ([4]C-NDTIs) triggers global aromaticity, yielding radical species with notable stability and optical properties.