Charge-Promoted Adaptive Aromaticity in Metallabenzenes: A Combined DFT and Machine Learning Study
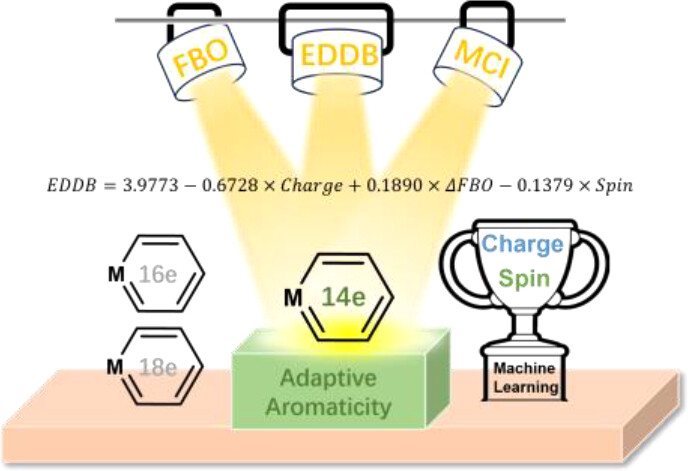
In accordance with the constraints by Hückel’s and Baird’s rules, species generally exhibit aromaticity in one state (the lowest singlet state S0 or the lowest triplet state T1). Consequently, species with adaptive aromaticity (being aromatic in both the S0 and T1 states) are particularly rare. In this study, density functional theory (DFT) was employed to investigate adaptive aromaticity in 14e–, 16e– and 18e– metallabenzenes. Based on bond length, fuzzy bond order (FBO), multicenter index (MCI), delocalized bond electron density (EDDB) analyses, magnetically induced current-density (MICD) analysis, and isomerization stabilization energy (ISE), 14e– ruthenabenzenes are found to possess adaptive aromaticity. A reduction in the delocalization of the spins in the six-membered ring of metallabenzenes will facilitate the maintenance of their T1 aromaticity. The ligand’s effect on adaptive aromaticity in 14e– ruthenabenzenes was conducted by principal component analysis (PCA), one of the most commonly used unsupervised machine learning algorithms, which suggests that electron-deficient ligands are detrimental to adaptive aromaticity. A supervised machine learning method, multiple linear regression (MLR), was employed to develop a quantitative equation for predicting aromaticity in the T1 state. This study demonstrates the first example of a metallabenzene with adaptive aromaticity, which is a valuable contribution to the field of aromatic chemistry.