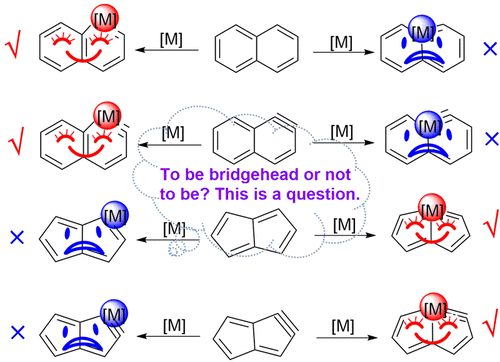
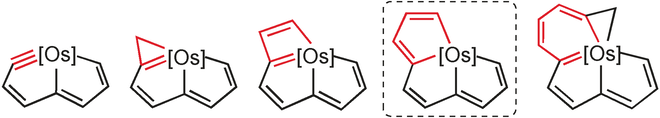
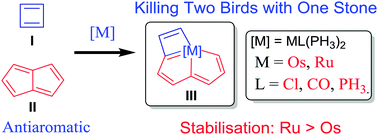
Releasing Antiaromaticity in Metal-Bridgehead Naphthalene
Submitted by Jun Zhu on Wed, 09/29/2021 - 15:34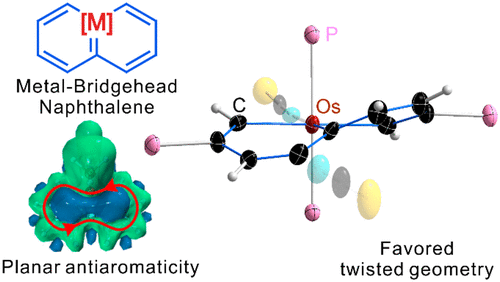
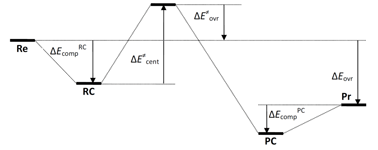
As a fundamental chemical property, aromaticity guides the synthesis of novel structures and materials. Replacing the carbon moieties of aromatic hydrocarbons with transition metal fragments is a promising strategy to synthesize intriguing organometallic counterparts with a similar aromaticity to their organic parents. However, since antiaromaticity will endow compound instability, it is a great challenge to obtain an antiaromatic organometallic counterpart based on such transition metal replacement in aromatic hydrocarbons.