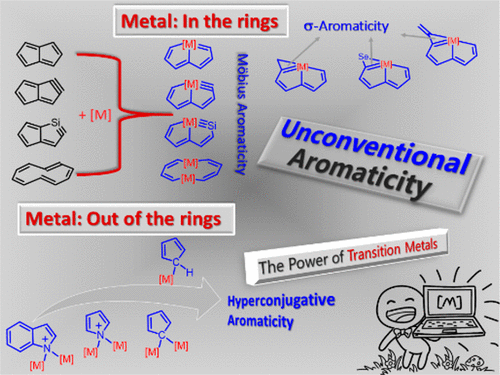
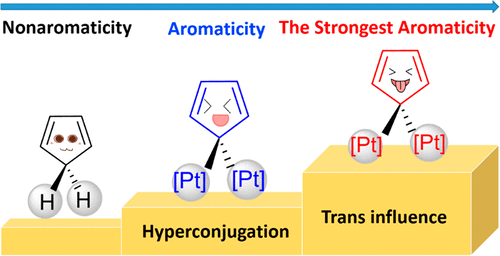
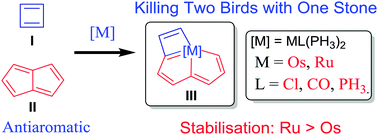
Access to tetracyclic aromatics with bridgehead metals via metalla-click reactions
Submitted by Jun Zhu on Sat, 02/29/2020 - 15:19
The never-ending pursuits for exploring aromatic molecular architectures result in the large libraries of aromatics with fascinating structures, which have greatly broadened the scope of aromaticity. Despite extensive efforts that have been paid to develop aromatic frameworks, the construction of polycyclic aromatics that share a bridgehead atom with more than three rings has never been accomplished.