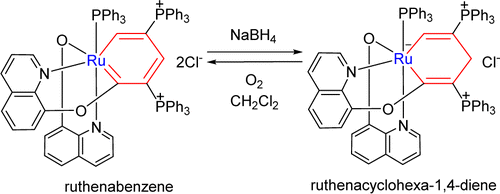
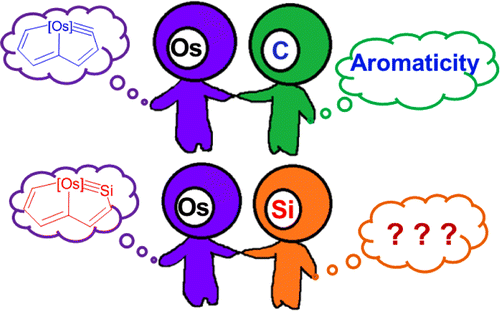
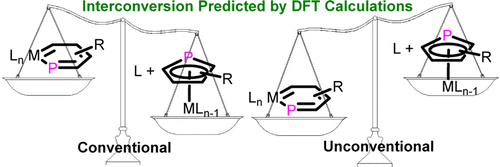
Probing the Strongest Aromatic Cyclopentadiene Ring by Hyperconjugation
Submitted by Jun Zhu on Wed, 09/12/2018 - 07:51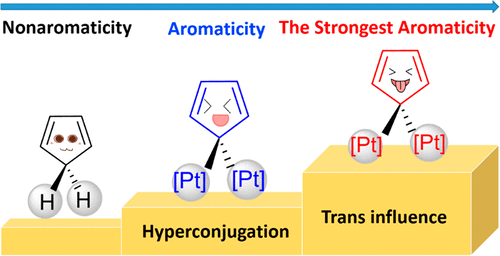
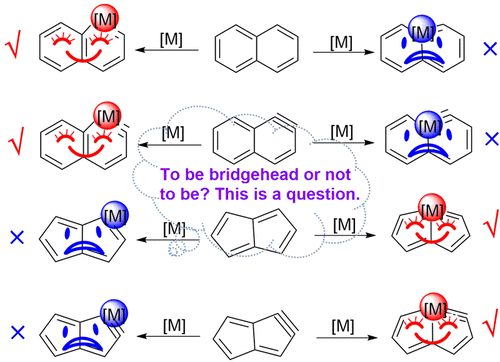
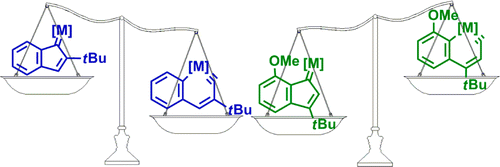
Hyperconjugation, an interaction of electrons in a σ orbital or lone pair with an adjacent π or even σ antibonding orbital, can have a strong effect on aromaticity. However, most work on hyperconjugative aromaticity has been limited to main-group substituents. Here, we report a thorough density functional theory study to evaluate the aromaticity in various cyclopentadienes that contain both main-group and transition-metal substituents.