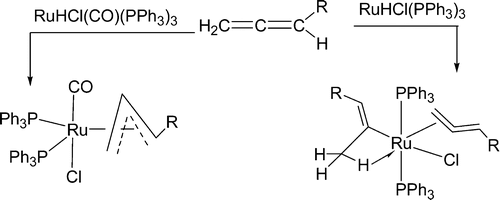
Insertion reactions of allenes with palladium aryl complexes [PdI(Ph)(PPh3)](2) and PdI(Ph)(dppe)
Submitted by Jun Zhu on Fri, 11/01/2013 - 00:06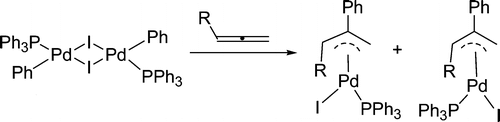
Treatment of [PdI(Ph)(PPh3)]2 with allenes CH2═C═CHR (R = CMe3, CO2Et, P(O)(OEt)2, and SO2Ph) in dichloromethane at room temperature produces a mixture of cis and trans isomers of the π-allyl palladium complexes PdI(η3-CH2C(Ph)CHR)(PPh3) in which the R group is anti to the Ph group. The disubstituted allenes MeCH═C═CHR (R = P(O)(OEt)2 and SO2Ph) similarly react with [PdI(Ph)(PPh3)]2 to give the π-allyl palladium complexes PdI(η3-MeCHC(Ph)CHR)(PPh3) in which the R group is anti and the Me group is syn to the Ph group.