Metallapentalenofurans and Lactone-Fused Metallapentalynes
Submitted by Jun Zhu on Fri, 05/05/2017 - 09:35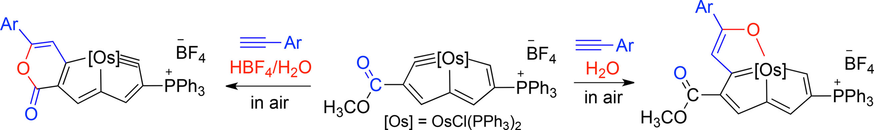
Metalla-aromatics are attractive species because they exhibit the properties of both organometallics and aromatics. Reported metal-bridged polycyclic aromatic complexes, as well as Möbius aromatic species, are still rare. Herein, we present the construction of two new metal-bridged polycyclic aromatic frameworks, α-metallapentalenofurans and lactone-fused metallapentalynes, by the reactions of osmapentalyne with terminal aryl alkynes in the presence of H2O or HBF4/H2O, respectively.