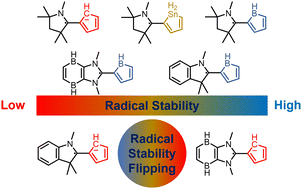
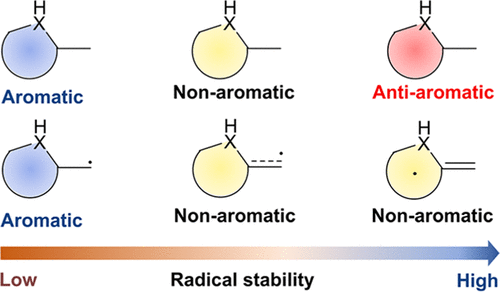
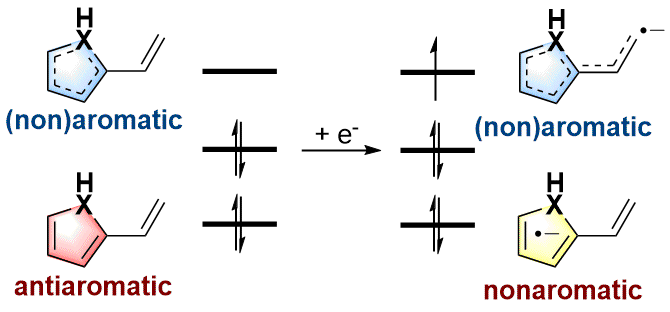Probing the longest λmax of azo compounds in near infrared absorption via integrating protonation, antiaromaticity and substituents: a combined DFT and machine learning study
Submitted by Jun Zhu on Mon, 01/26/2026 - 16:29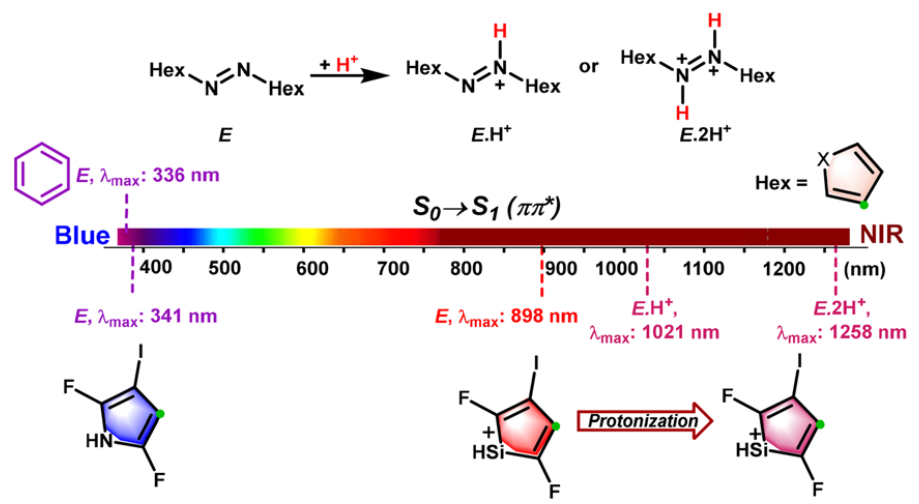
The photoisomerism of azo switches using light in the near-IR region (NIR, 780-1400 nm) is highly preferable for the applications to biomedical and pharmacological fields. The common chemical modifications of azobenzene only enables the E ⇆ Z photoswitching wavelengths of azobenzene derivatives close to the red limit of near-infrared light.