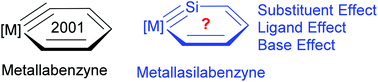
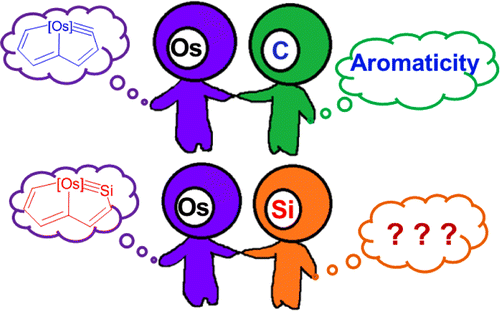
Nature Chemistry
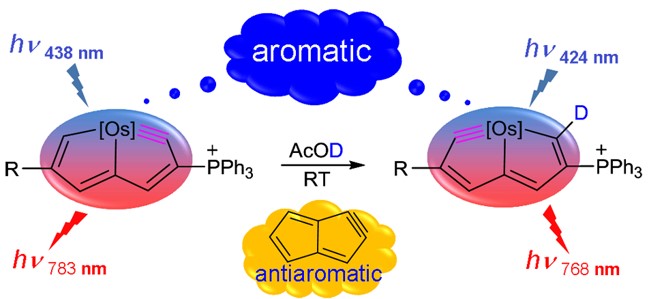
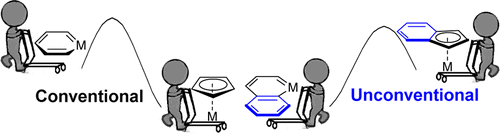
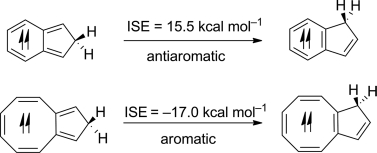
Pentalyne — a bicyclic anti-aromatic molecule that contains a strained five-membered ring — is a challenging synthetic target. Incorporation of an osmium centre has been shown to stabilize such features and enable the preparation of two osmapentalyne derivatives. Despite featuring the smallest angles observed so far at a carbyne moiety, these molecules benefit from Möbius aromaticity and reduced ring strain.